
By 2025, the global advanced therapy medicinal products (ATMP) sector, encompassing cell therapies, gene therapies, and genetically modified cell-based medicines, has progressed from early scientific success to clinical and commercial reality. Following a decade of rapid innovation, the ATMP sector is now characterised by a growing number of approved therapies, expanding therapeutic indications, and increasing global competition. The patient benefits of ATMPs are undeniable with a year-on-year growth in the number of patients who are benefitting from advanced therapies globally. Patients who previously had no treatment options are now being offered renewed hope through new innovative ATMPs. At the same time, the sector has faced commercial hurdles in 2025 including manufacturing and supply chain challenges, pricing and reimbursement scrutiny, and a renewed emphasis on demonstrating sustainable clinical and economic value at scale.
In 2025, 20 of the 30 largest biopharma companies by market cap are now investing in the development and commercialisation of ATMPs. Research conducted by Citeline reported ATMP sector funding of $11.1 Billion in 2025. The ATMP sector comprised approximately 18% of the value of all biotech venture financing in 2025, an increase in the reported 15% value report in 2024.
The Race to the Clinic: Asia Pacific Region Surpasses North America
In the past year, the Asia-Pacific region overtook North America for the first time in the number of ATMP clinical trials, signalling a significant shift in the global advanced therapy landscape. There were an estimated 916 clinical trials ongoing in North America in 2025, with 890 of these in the United States, while the Asia-Pacific region conducted approximately 990 trials, 716 of them in China. This growth in the Asia-Pacific region reflects the region’s growing focus on accelerating the translation of novel therapies from lab to clinic, supported by evolving government policies that are driving faster initiation of in-human studies. The trend highlights a broader intensifying competition to bring breakthrough ATMPs to patients worldwide.
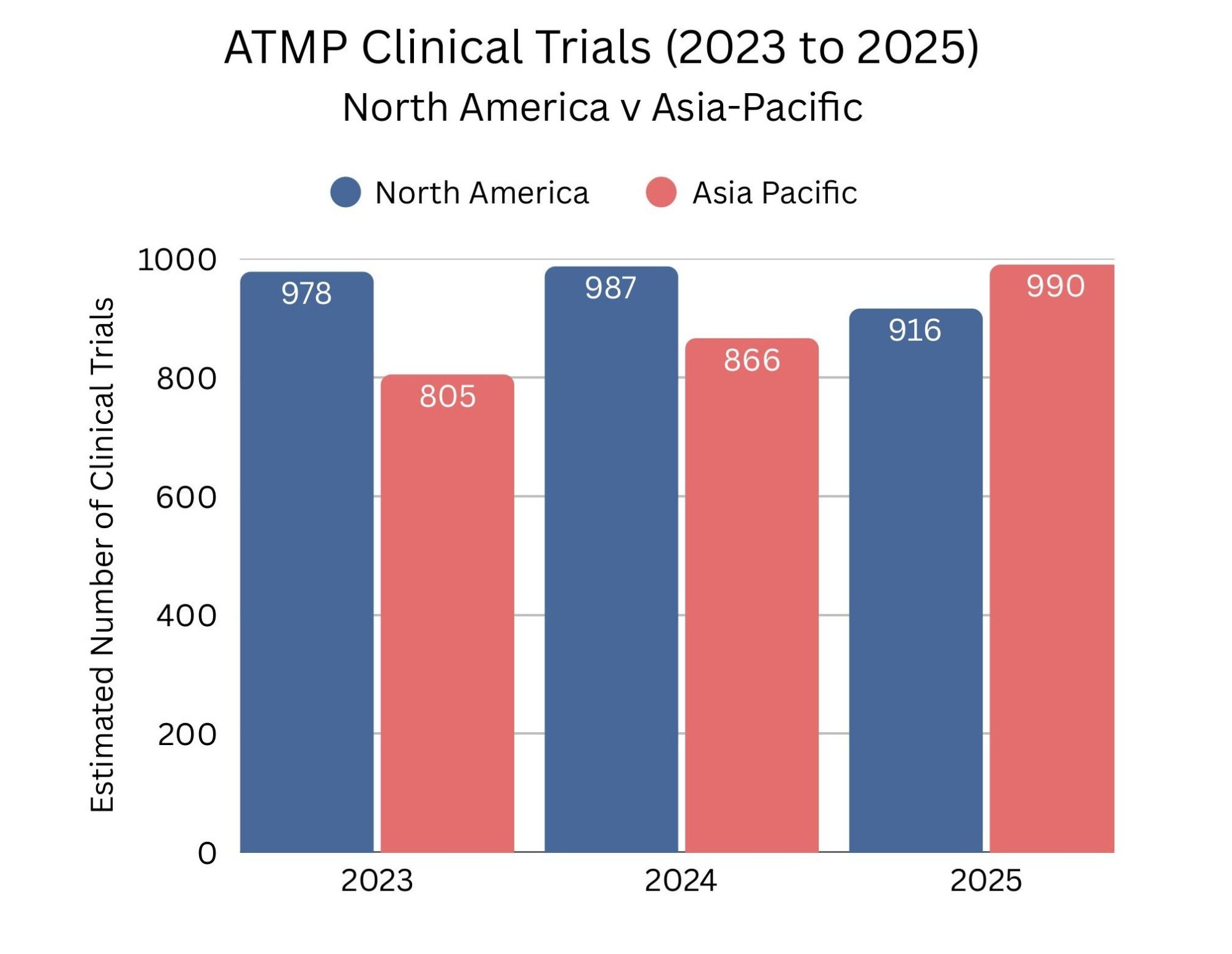
Source data: www.alliancerm.org
FDA and EMA Approvals in 2025
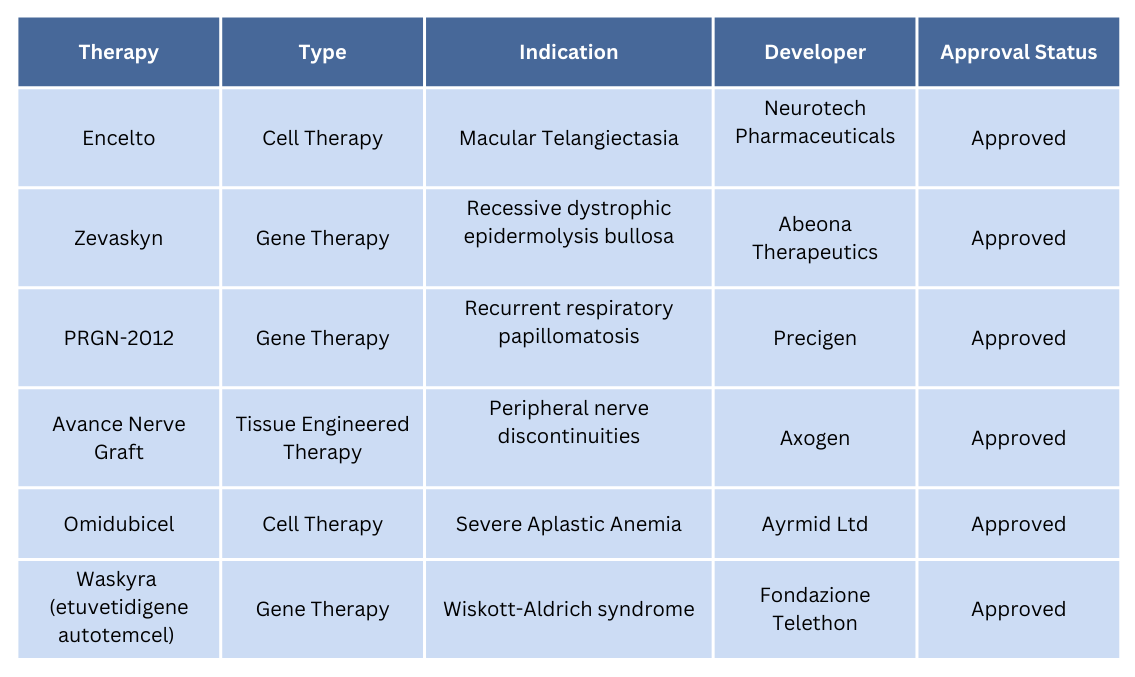
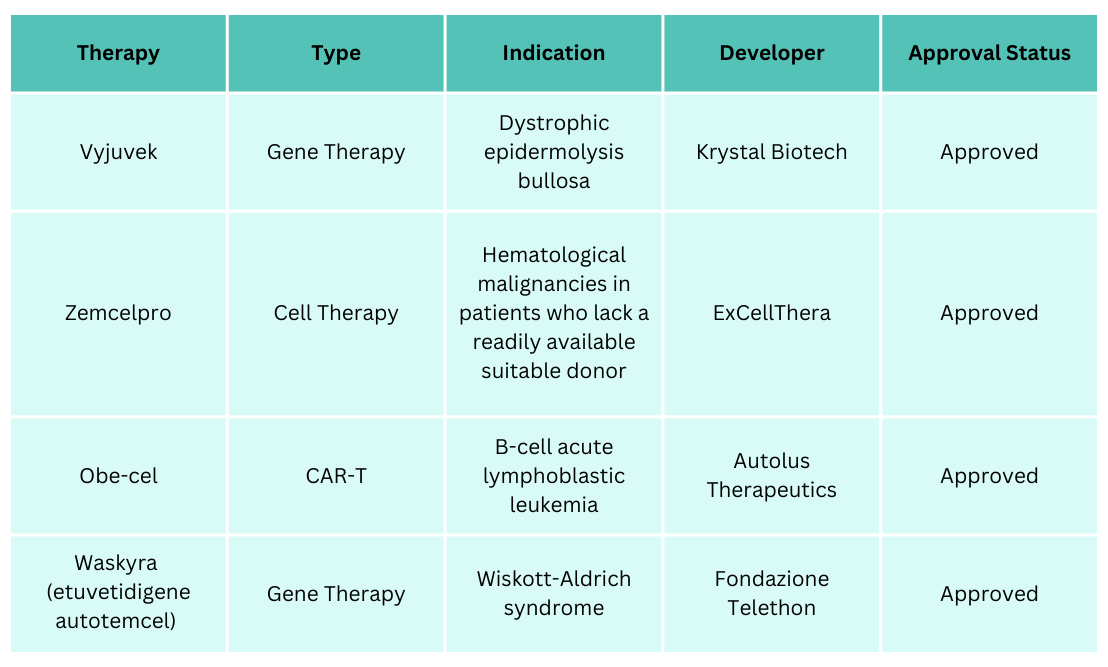
Over the past year, both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) granted several significant approvals for ATMPs. In total, the FDA approved six ATMPs, while the EMA approved four. A major milestone in 2025 was the approval of Fondazione Telethon’s etuvetidigene autotemcel (Waskyra) for Wiskott–Aldrich syndrome (WAS) by both the FDA and EMA. Waskyra is the first gene therapy approved for this rare disease, and Fondazione Telethon became the first non-profit organisation to achieve gene therapy approval. The tables below provide details on these newly authorised ATMPs.
ATMPs that were approved by the FDA in 2025
ATMPs that were approved by the EMA in 2025
Mesoblast’s Ryoncil became commercially available in the US in March 2025
At the end of 2024, Melbourne-based biotech Mesoblast secured FDA approval for Ryoncil, the first mesenchymal stromal cell (MSC) therapy to be approved by the FDA for any indication. Ryoncil is now an approved treatment for children aged two months and older, including adolescents, with steroid-refractory acute graft-versus-host disease (SRaGvHD), a life-threatening condition associated with high mortality. The therapy became commercially available in the US on 28th March 2025. Sales of Ryoncil reached US$13.2 million in the second quarter of 2025, increased to US$21.9 million in the third quarter (a 66% quarter-on-quarter increase), and rose further to US$35.1 million in the fourth quarter (approximately 60% growth over the previous quarter). This accelerating commercial performance reflects the growing number of patients benefiting from this MSC-based therapy.
Significant Growth in Outpatient CAR-T Treatment
Among the positive developments has been the continued expansion of ATMP treatment access in outpatient settings. Outpatient CAR-T treatment has experienced significant growth in recent years, reflecting increasing understanding and clinical education on CAR-T administration, increased safety profiles, improved toxicity management and adapting healthcare infrastructure. Continuous improvements by developers and healthcare teams have made it possible to safely transition appropriate patients from inpatient to outpatient settings. Standardised monitoring protocols, remote symptom tracking, and rapid-response care pathways have increased clinician confidence in outpatient delivery while reducing hospital length of stay and overall treatment costs. This trend has enhanced patient convenience and quality of life, eased capacity constraints in hospitals, and positioned outpatient CAR-T therapy as an increasingly viable treatment option for some patients.
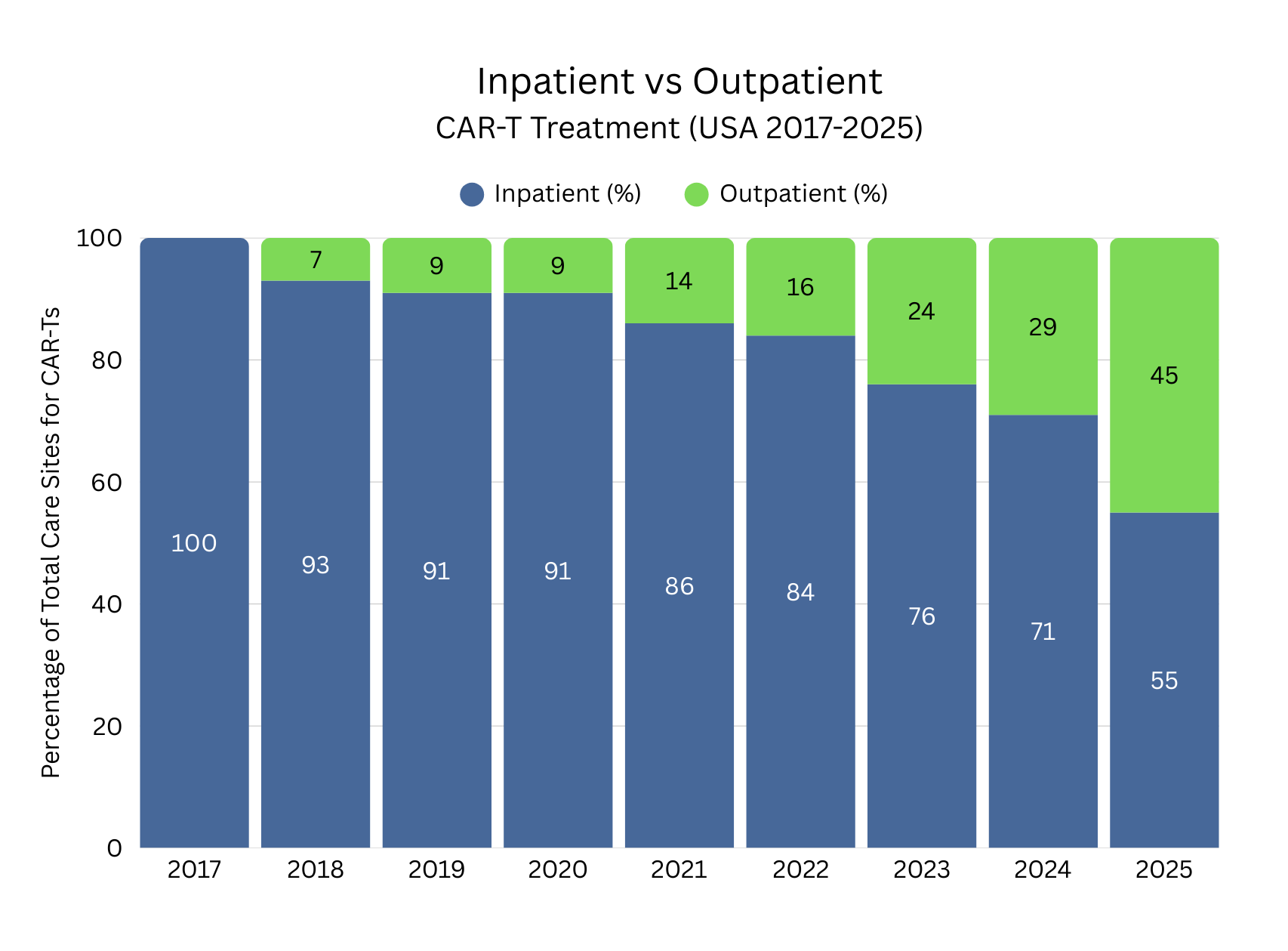
Source data: Guidehouse Analysis of McKesson Compile Patient Ready Data (US)
European Commission Proposes Biotech Act to Strengthen EU Health Biotechnology
The European Commission published its proposal for a European Biotech Act in December 2025, as outlined in its 2024–2029 Political Guidelines. The Act seeks to create a supportive framework to accelerate the development of biotechnology products from research to market, while maintaining high safety and ethical standards. It represents a potential important milestone for ATMPs in Europe by seeking to better align regulation, investment, and patient access.
The Act combines regulatory improvements, targeted incentives, and funding measures to boost clinical trial activity, support ATMP centres of excellence, and enhance intellectual property protection, including a 12-month extension of supplementary protection certificates. It also aims to address Europe’s long-standing challenges in translating scientific excellence into commercial success, particularly in health biotechnology, where complex legislation and funding gaps had led to many developers focusing on other markets such as North America
Here are three main takeaways from the recently adopted Act:
- The Biotech Act seeks to address structural barriers limiting Europe’s ability to commercialise biotech innovation, including funding constraints and a declining share of global clinical trials.
- It introduces coordinated measures across seven core pillars, including strategic project designations, fast-track permitting, additional funding, and extended SPCs for qualifying biotech and ATMP products.
- The Act streamlines existing EU regulatory frameworks (including CTR, ATMP, and SoHO) through accelerated timelines, risk-based requirements, and regulatory sandboxes, aiming to reduce time to market while preserving high safety standards.
In parallel with the Biotech Act proposal, the Commission and the European Investment Bank Group announced BioTechEU, an initiative to mobilise €10 billion in public-private investment for the biotech and life sciences sectors in the next year.
ATMP Companies are Adapting Fast
To address the challenges within the ATMP sector, companies are increasingly required to adapt quickly and operate with greater agility in a rapidly evolving landscape. A growing number of developers are focusing on delivering best-in-class advanced therapies for indications with high unmet need and significant patient populations, including Parkinson’s disease, Multiple myeloma, Huntington’s disease, and others. This shift reflects both scientific progress and a broader commitment to addressing diseases where conventional treatment options remain limited.
In the early days of the ATMP sector, access to treatment was constrained by significant operational and infrastructure barriers, including a limited number of specialised treatment centres, Today, the sector is actively working to dismantle these barriers and expand patient access. For example, Carvykti, a personalised CAR-T cell therapy for adults with relapsed or refractory multiple myeloma, is now administered in an outpatient setting approximately 50% of the time. Developers are collaborating closely with regulatory agencies to enhance safety profiles and refine clinical trial designs in response to regulatory feedback, supporting more efficient and patient-centric development pathways. Heading into 2026, a significant number of regulatory decisions and submissions are planned for first half of the year and global clinical trials are gaining momentum.
Speak with Our Team
HiTech Health is a leading European CDMO and service provider for ATMPs. We deliver end-to-end solutions from preclinical development through to GMP manufacturing and global patient supply. If you require support with the development and manufacturing of an ATMP, then contact us today.
Author: Paul Crozier – January 2026
References:
www.alliancerm.org/resources/?_resource_type=cell-gene-therapy-sector-data
https://alliancerm.org/wp-content/uploads/2026/01/SOTI-2026-Industry-Update.pdf
www.citeline.com/en/resources/q3-2025-gene-cell-and-rna-therapy-report
https://www.eesc.europa.eu/en/our-work/opinions-information-reports/opinions/biotech-act