by Paul Crozier | Jul 21, | HiTech Health Latest News
For companies manufacturing or importing medicinal products into the EU or UK, the role of the Qualified Person (QP) is a mandatory regulatory requirement. What exactly is a QP, and why is this role so important?
What is the Role of a Qualified Person?
A Qualified Person (QP) is a highly trained and experienced professional who is legally responsible for ensuring that medicinal products meet all required quality, safety, and regulatory standards before they are released to the market or for use in clinical trials.
The QP must certify that each batch of a medicinal product has been manufactured and tested in accordance with Good Manufacturing Practice (GMP), the product’s Marketing Authorisation/Clinical Trial Authorisation, and all applicable regulations.
Why is a QP Required?
The QP requirement was established to protect patients and maintain confidence in the supply chain of medicinal products. Before any medicinal product can be distributed in the EU or UK, a QP must formally certify that the batch complies with regulatory requirements.
This independent oversight helps to:
- Ensure patient safety
- Verify compliance with GMP regulations
- Confirm product quality and consistency
- Support regulatory inspections and audits
- Reduce the risk of defective or non-compliant products reaching the market
When is QP Certification Needed?
QP certification is required for:
- Commercial (‘to-market’) batch release of medicinal products:
- Manufactured within the EU or UK
- Imported from countries outside the EU or UK
- Investigational Medicinal Products (IMPs) used in clinical trials
Without QP certification, medicinal products cannot legally be released for sale or supply within the relevant market or for use in clinical trials.
Can I Outsource the QP requirement?
Many pharmaceutical and biotechnology companies choose to outsource QP responsibilities to experienced consultants or specialist service providers like HiTech Health. External organisations can support organisations with batch certification, GMP compliance, quality oversight, regulatory inspections, and supply chain management. However, it is important to do a thorough due diligence and select an experienced and trusted provider.
By partnering with an experienced company like HiTech Health, companies can ensure compliance with complex regulatory requirements while maintaining efficient product release timelines. The Qualified Person plays a vital role in safeguarding the quality and safety of medicinal products across the EU and UK. Whether for routine batch release, product importation, or clinical trial supply, QP certification is a legal requirement that helps ensure patients receive medicines that meet the highest standards of quality and compliance. We can help with all quality and compliance activities from your overall quality management system through quality audits and QMS remediation support.
Contact our team today and schedule a call to learn more about our QP services for the EU and UK markets.
by Paul Crozier | Jun 29, | HiTech Health Latest News
Successfully conducting a clinical trial in the UK extends beyond developing an Investigational Medicinal Product (IMP) and recruiting participants. Sponsors must also establish robust regulatory, quality, and operational processes to safeguard participant safety, ensure the integrity of clinical data, and comply with Medicines and Healthcare products Regulatory Agency (MHRA) requirements. Understanding these requirements early can help avoid costly delays and support a successful clinical development programme.
Regulatory Approval
Before a clinical trial can begin, sponsors must obtain the necessary approvals from the relevant authorities. These typically include:
- Clinical Trial Authorisation (CTA) from the MHRA
- Approval from a Research Ethics Committee (REC)
- Site-specific approvals and contracts where required
The application process requires detailed information on the investigational medicinal product, study protocol, investigator brochure, and supporting quality documentation.
Investigational Medicinal Product (IMP) Manufacturing
Any investigational medicinal product used in a clinical trial must be manufactured in accordance with Good Manufacturing Practice (GMP). This includes:
- Manufacturing under an appropriate GMP licence
- Validated manufacturing and testing processes
- Quality control testing
- Stability studies
- Comprehensive batch documentation that will be reviewed by a Qualified Person (QP)
Maintaining GMP compliance throughout the manufacturing process is essential to ensure the quality, safety, and consistency of the IMP.
Qualified Person (QP) Certification
One of the most important requirements for clinical trials involving medicinal products is QP certification.
Before an IMP can be released for use in a UK clinical trial, a Qualified Person (QP) must certify that each batch has been manufactured and tested in accordance with GMP requirements, the clinical trial application, and applicable regulations.
QP certification provides an independent assessment of product quality and helps ensure that trial participants receive investigational products that meet the required standards.
IMP Importation and Supply Chain Management
Where investigational products are manufactured outside the UK, additional controls may be required to ensure compliance with UK regulations. Sponsors must consider:
- Importation requirements
- Supply chain oversight
- Temperature-controlled distribution
- Storage and accountability procedures
- Product traceability throughout the trial
A QP can play a key role in reviewing imported product documentation and supporting compliant batch release activities.
Ongoing Quality Oversight
Clinical trial compliance does not end once the study begins. Sponsors must maintain robust quality systems to manage:
- Deviations and non-conformances
- Product complaints
- Quality investigations
- Change controls
- Product recalls, if required
Experienced QPs can provide valuable support in assessing quality events, performing risk assessments, and ensuring regulatory expectations are met throughout the study lifecycle.
Why Engage a Qualified Person Early?
Many sponsors engage QP services at an early stage of development to avoid delays during clinical trial preparation and product release. Early QP involvement can help with:
- IMP release strategy
- GMP compliance assessments
- Batch certification
- Importation requirements
- Quality documentation review
- Investigation and recall support
- Regulatory inspection readiness
By incorporating QP expertise into the clinical trial programme from the outset, sponsors can reduce compliance risks and maintain confidence in their product supply chain.
Conclusion
Successfully conducting a clinical trial in the UK requires careful planning, robust quality systems, and compliance with regulatory requirements. Qualified Person services play a critical role in ensuring investigational medicinal products are manufactured, assessed, and released in accordance with GMP and clinical trial regulations.
Whether supporting IMP certification, importation activities, or ongoing quality oversight, an experienced QP provider like HiTech Health can help sponsors navigate the complexities of clinical trial compliance and keep development programmes moving forward.
Contact our team today and schedule a call to learn more about our services for the EU and UK markets.
by Paul Crozier | Jun 22, | HiTech Health Latest News
Research Ireland has launched the RINN national research network, a landmark €460 million investment aimed at strengthening Ireland’s research ecosystem and accelerating innovation across strategic sectors including Advanced Therapies, AI, Quantum Technologies, Energy, Semiconductors, Pharma & Biopharma, and Medical Devices.
A key pillar is RINN Advanced Therapies, a national integrated research centre focused on developing, manufacturing, and clinically translating personalised advanced cellular immune therapeutics (ACITs). These next-generation cell and immune-based medicines are expected to play a growing role in treating cancer, autoimmune disease, and other complex conditions. HiTech Health welcomes this significant announcement and looks forward to seeing the continued growth of the ecosystem.
A National Ecosystem For Cell Therapy Innovation
RINN Advanced Therapies brings together a broad consortium of Irish universities, hospitals, research institutes, and national bodies to create an all-island innovation ecosystem. Partners include Trinity College Dublin, University of Galway, UCC, Maynooth University, TU Dublin, RCSI, along with translational partners such as NIBRT and key health system stakeholders.
The centre aims to bridge gaps across the advanced therapies pipeline by connecting discovery, biomanufacturing, regulatory science, and clinical delivery into a coordinated framework.
From Discovery to Patient Impact
The programme focuses on accelerating translation of cell and immune-based treatments into clinical use by integrating:
- Advanced cell therapy design and engineering
- Biomanufacturing and scale-up of personalised therapies
- Clinical translation and patient-centred delivery models
- Regulatory and quality frameworks for novel biologics
This approach aims to enable more personalised therapies with improved outcomes, particularly in oncology and immune-mediated diseases.
Positioning Ireland globally
Advanced therapies are one of the fastest-growing areas in biopharma, driven by progress in cell and gene therapy, immunotherapy, and precision medicine. RINN Advanced Therapies strengthens Ireland’s position by linking research excellence with industrial capability. By aligning academia, healthcare, and biomanufacturing, the network will support a sustainable innovation pipeline that advances both scientific leadership and patient impact.
Looking ahead
Over the coming years, RINN Advanced Therapies will act as a national platform for collaboration, training, and innovation, supporting the next generation of researchers and clinicians while accelerating delivery of transformative therapies. As part of the wider RINN network, it marks a significant step toward a more coordinated, impact-driven research system focused on translating discovery into real health outcomes.
Speak with our team today
by Paul Crozier | May 25, | HiTech Health Latest News
Artificial Intelligence (AI) is rapidly becoming an important tool across the pharmaceutical and biotechnology sectors, with applications ranging from process monitoring and predictive analytics to deviation detection and classification. As AI adoption accelerates by life sciences companies, regulators are increasingly focused on ensuring AI technologies are implemented in a controlled, effective, and GMP-compliant manner.
The European Commission has recently issued a draft version of EU GMP Annex 22, a new guideline specifically addressing the use of AI in GMP-regulated manufacturing environments. The proposed annex is intended to complement existing requirements under Annex 11 for computerised systems and introduces a risk-based framework for AI deployment in pharmaceutical manufacturing.
The draft guidance applies to AI and machine learning (AI/ML) systems used in critical GMP applications that may impact patient safety, product quality, or data integrity. The current draft guideline focuses on ‘static’ AI models; models that do not adapt their performance during use by incorporating new data. The draft Annex 22 states that ‘dynamic’ models that continuously learn and adapt during use are outside its scope and should not be used in critical GMP applications.
Several key themes emerge throughout Annex 22:
- Clearly defining the intended use of AI models
- Robust validation and acceptance criteria
- Independence and quality of test datasets
- Explainability and transparency of AI outputs
- Human oversight and governance
- Ongoing monitoring and lifecycle management
The guidance also highlights the importance of cross-functional collaboration between quality, manufacturing, IT, process SMEs, and data scientists throughout the AI lifecycle.
For companies operating in the cell and gene therapy sector, these developments are particularly significant. Advanced therapy manufacturing generates large and complex datasets, making AI an increasingly attractive tool for process optimisation, analytics, and operational efficiency. However, the regulatory expectations surrounding validation, data governance, and explainability will require careful consideration as AI adoption expands.
While Annex 22 remains in draft form, it provides a strong indication of the future regulatory direction for AI within GMP manufacturing environments. Organisations that begin establishing appropriate governance frameworks, validation strategies, and quality oversight processes today will be better positioned for future compliance and successful implementation of AI-enabled technologies.
HiTech Health is a leading provider of QP services in the EU and the UK. Our QP team have extensive knowledge of pharmaceuticals, biologics and Advanced Therapeutic Medicinal Products (ATMPs) including cell and gene therapies. We can help with all quality and compliance activities from your overall quality management system through quality audits and QMS remediation support.
Contact our team today and schedule a call to learn more about our services for the EU and UK markets.
References:
https://health.ec.europa.eu/document/download/5f38a92d-bb8e-4264-8898-ea076e926db6_en?filename=mp_vol4_chap4_annex22_consultation_guideline_en.pdf
by Paul Crozier | Apr 22, | HiTech Health Latest News
The success of a cell therapy extends well beyond development and GMP manufacturing. It also depends on the ability to deliver the product safely, reliably, and under tightly controlled conditions. Unlike traditional pharmaceuticals, cell therapies are highly sensitive, patient-specific, and of very high value. As a result, cold chain logistics is not just a supporting function, it is fundamental to maintaining cell viability and ensuring successful patient outcomes.
In this article, HiTech Health explores the key risks associated with cell therapy logistics and outlines considerations for managing these risks from manufacturing facility to patient.
The Unique Challenge of Cell Therapy Logistics
Cell therapies present a level of logistical complexity far beyond conventional medicines. These products are typically cryopreserved at ultra-low temperatures (≤ -150°C), often time-sensitive, and, in many cases, patient-specific, requiring full traceability at every stage. Cell therapies are also frequently transported across international borders, adding further regulatory, customs and operational complexity. Any deviation in temperature, delays in transit, or breakdown in chain of identity can compromise product integrity and ultimately impact patient outcomes.
Managing Risk in the Cold Chain
One of the most critical risks with shipping cell therapies is temperature excursion. Maintaining ultra-low temperatures throughout transit is essential, as even brief deviations can affect cell viability and potency. This risk is often heightened by delays at customs, the use of inexperienced transportation providers, or inadequately qualified shipping lanes or equipment. Mitigation strategies include the use of validated cryoshippers with proven hold times, pre-qualified shipping lanes, and continuous temperature monitoring with real-time visibility.
Equally important is maintaining a robust chain of identity and custody, particularly for autologous therapies. Any mismatch between patient and product is unacceptable. This risk can be effectively managed through digital tracking systems, secure labelling solutions such as barcoding or RFID, and comprehensive documentation across the entire supply chain.
Operational variability also introduces risk. Cell therapies typically move through multiple stakeholders, including manufacturing sites, storage depots, and clinical sites. Each handoff creates potential for error. Standardised handling procedures, well-trained logistics partners, and clear communication pathways are essential to ensure consistency and control. Teams should also consider implementing documentation checklists to prevent delays in exportation and importation processes.
Regulatory compliance adds another layer of complexity. International shipments must navigate differing import/export and customs requirements. Early planning, accurate documentation, and collaboration with experienced partners are key to avoiding delays and ensuring compliance, so that patients can receive their treatments in a timely manner. For therapies supplied in the EU and UK markets, having a Manufacturer’s/Importer’s Authorisation (MIA) holder and an experienced Qualified Person (QP) team is critical to reviewing the cold chain from a quality and compliance perspective.
Building a Resilient Cold Chain Strategy
A robust cold chain strategy requires a proactive, end-to-end approach. This includes qualifying shipping routes under real-world conditions, conducting thorough risk assessments, and implementing contingency plans for potential disruptions such as delays or equipment failures.
Efficient cold chain logistics is not simply about transporting a cell therapy, it is about protecting the integrity of a potentially life-saving treatment at every stage of its journey. By understanding the unique risks involved and implementing effective mitigation strategies, developers can improve reliability, reduce failure rates, and ensure that high quality therapies reach patients on time for their treatment.
HiTech Health has extensive experience supporting developers in implementing robust supply chains for cell therapies, with an in-house team of GDP-trained supply chain personnel. The company also holds MIAs in both the EU and UK to support clinical trials and commercial launches.
Contact our team today and schedule a call to learn more about how we can support your cell therapy logistics requirements.
by Paul Crozier | Jan 22, | HiTech Health Latest News
By 2025, the global advanced therapy medicinal products (ATMP) sector, encompassing cell therapies, gene therapies, and genetically modified cell-based medicines, has progressed from early scientific success to clinical and commercial reality. Following a decade of rapid innovation, the ATMP sector is now characterised by a growing number of approved therapies, expanding therapeutic indications, and increasing global competition. The patient benefits of ATMPs are undeniable with a year-on-year growth in the number of patients who are benefitting from advanced therapies globally. Patients who previously had no treatment options are now being offered renewed hope through new innovative ATMPs. At the same time, the sector has faced commercial hurdles in 2025 including manufacturing and supply chain challenges, pricing and reimbursement scrutiny, and a renewed emphasis on demonstrating sustainable clinical and economic value at scale.
In 2025, 20 of the 30 largest biopharma companies by market cap are now investing in the development and commercialisation of ATMPs. Research conducted by Citeline reported ATMP sector funding of $11.1 Billion in 2025. The ATMP sector comprised approximately 18% of the value of all biotech venture financing in 2025, an increase in the reported 15% value report in 2024.
The Race to the Clinic: Asia Pacific Region Surpasses North America
In the past year, the Asia-Pacific region overtook North America for the first time in the number of ATMP clinical trials, signalling a significant shift in the global advanced therapy landscape. There were an estimated 916 clinical trials ongoing in North America in 2025, with 890 of these in the United States, while the Asia-Pacific region conducted approximately 990 trials, 716 of them in China. This growth in the Asia-Pacific region reflects the region’s growing focus on accelerating the translation of novel therapies from lab to clinic, supported by evolving government policies that are driving faster initiation of in-human studies. The trend highlights a broader intensifying competition to bring breakthrough ATMPs to patients worldwide.
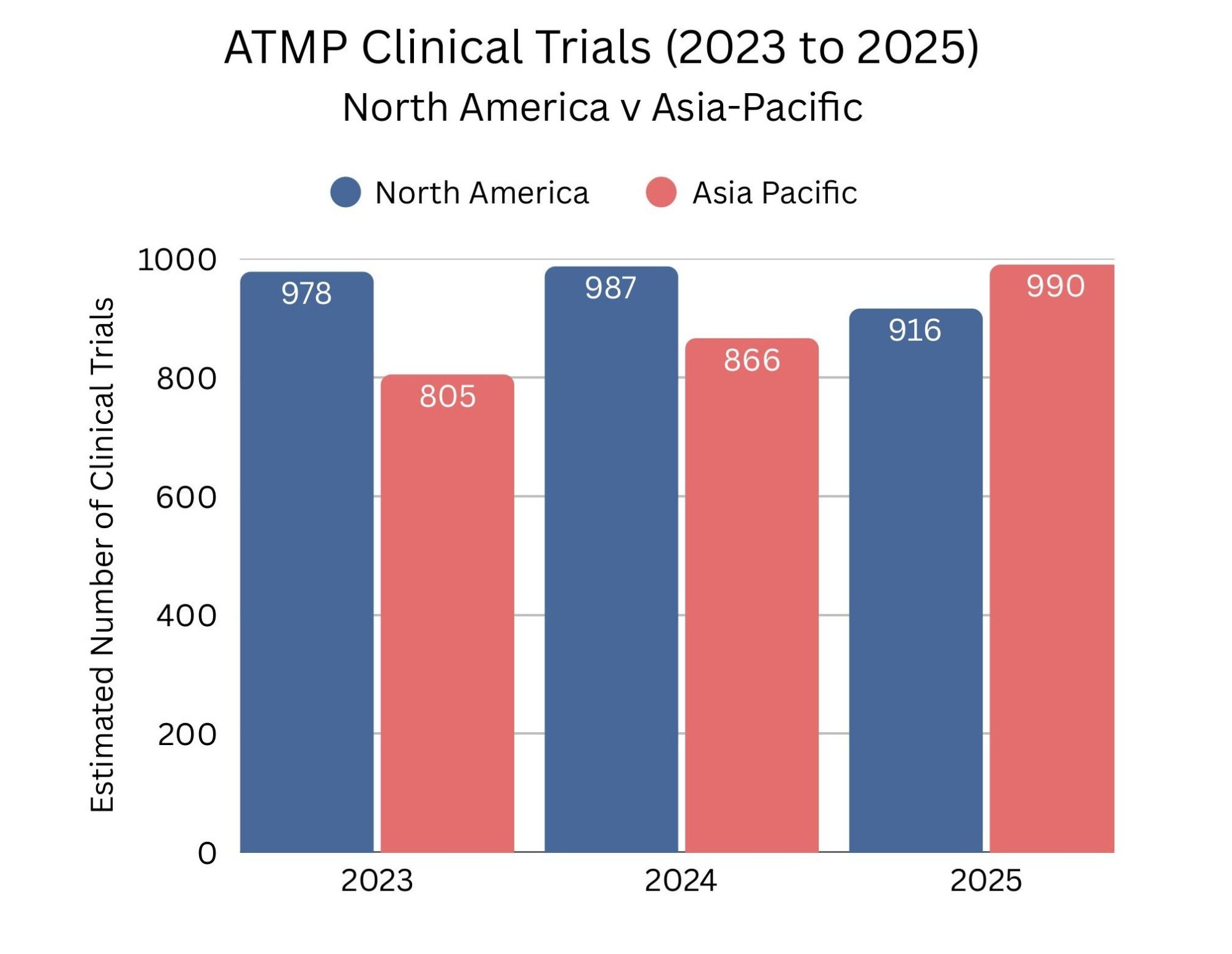
Source data: www.alliancerm.org
FDA and EMA Approvals in 2025
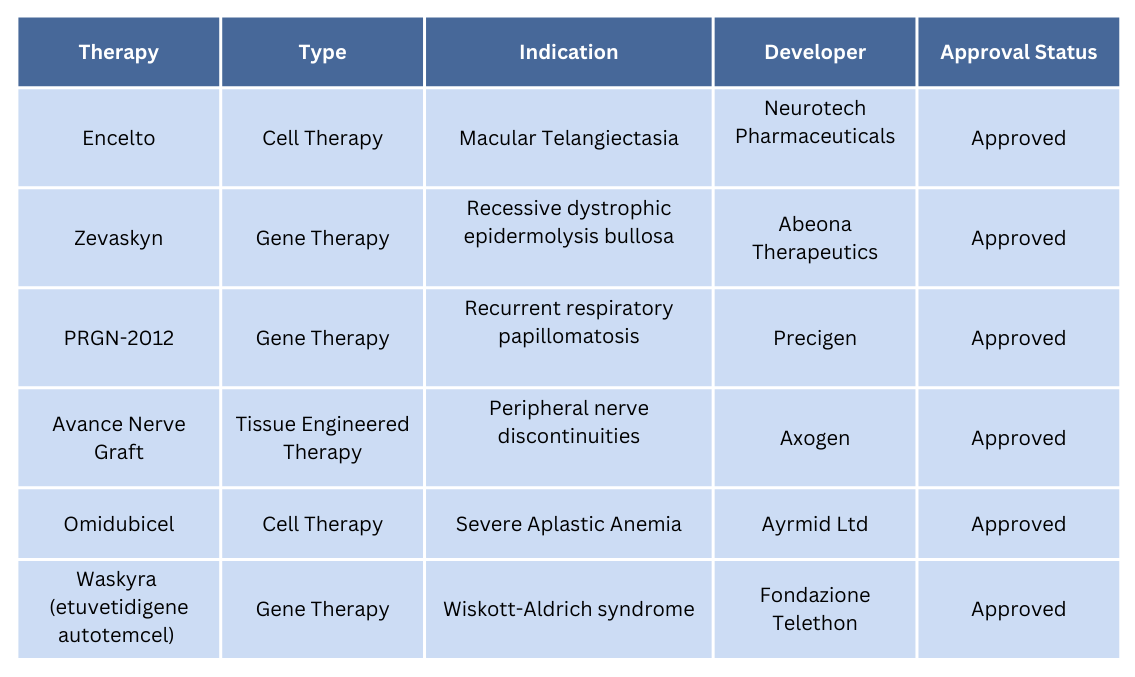
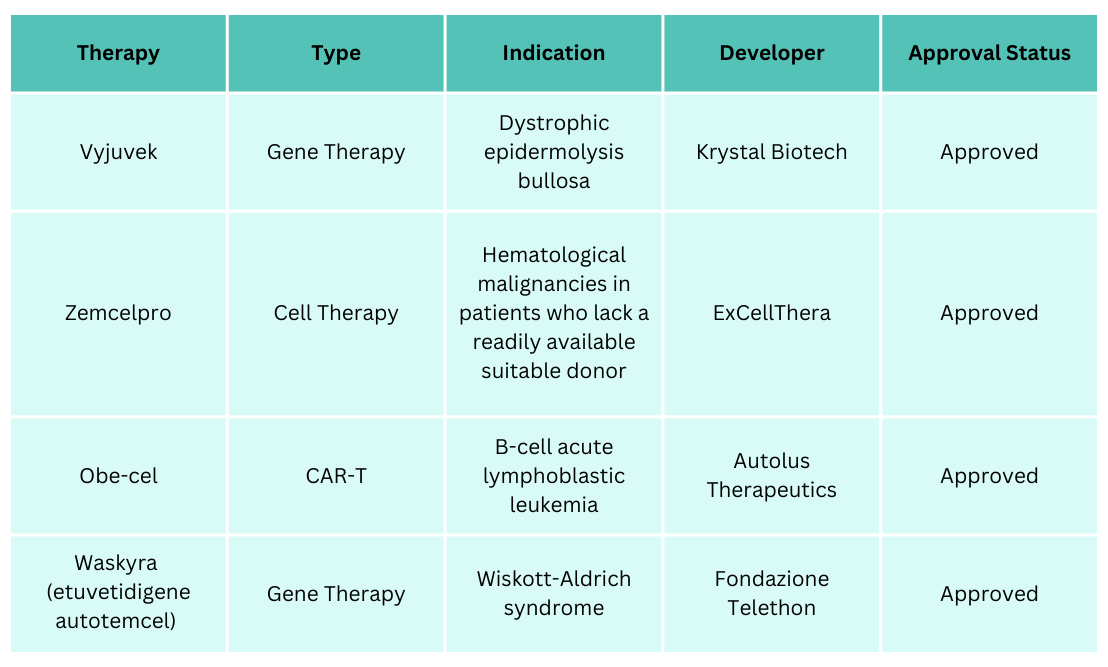
Over the past year, both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) granted several significant approvals for ATMPs. In total, the FDA approved six ATMPs, while the EMA approved four. A major milestone in 2025 was the approval of Fondazione Telethon’s etuvetidigene autotemcel (Waskyra) for Wiskott–Aldrich syndrome (WAS) by both the FDA and EMA. Waskyra is the first gene therapy approved for this rare disease, and Fondazione Telethon became the first non-profit organisation to achieve gene therapy approval. The tables below provide details on these newly authorised ATMPs.
ATMPs that were approved by the FDA in 2025
ATMPs that were approved by the EMA in 2025
Mesoblast’s Ryoncil became commercially available in the US in March 2025
At the end of 2024, Melbourne-based biotech Mesoblast secured FDA approval for Ryoncil, the first mesenchymal stromal cell (MSC) therapy to be approved by the FDA for any indication. Ryoncil is now an approved treatment for children aged two months and older, including adolescents, with steroid-refractory acute graft-versus-host disease (SRaGvHD), a life-threatening condition associated with high mortality. The therapy became commercially available in the US on 28th March 2025. Sales of Ryoncil reached US$13.2 million in the second quarter of 2025, increased to US$21.9 million in the third quarter (a 66% quarter-on-quarter increase), and rose further to US$35.1 million in the fourth quarter (approximately 60% growth over the previous quarter). This accelerating commercial performance reflects the growing number of patients benefiting from this MSC-based therapy.
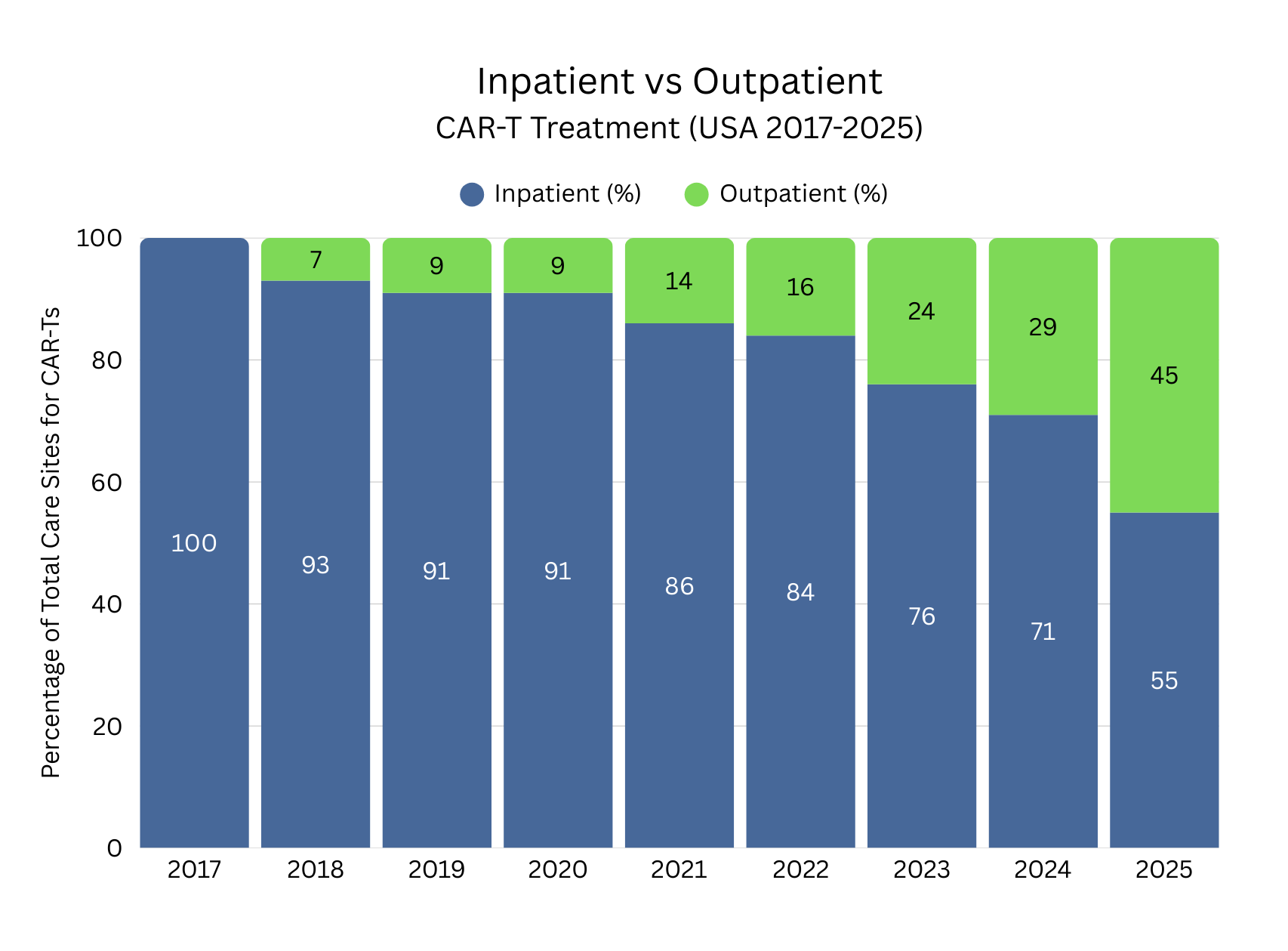
Significant Growth in Outpatient CAR-T Treatment
Among the positive developments has been the continued expansion of ATMP treatment access in outpatient settings. Outpatient CAR-T treatment has experienced significant growth in recent years, reflecting increasing understanding and clinical education on CAR-T administration, increased safety profiles, improved toxicity management and adapting healthcare infrastructure. Continuous improvements by developers and healthcare teams have made it possible to safely transition appropriate patients from inpatient to outpatient settings. Standardised monitoring protocols, remote symptom tracking, and rapid-response care pathways have increased clinician confidence in outpatient delivery while reducing hospital length of stay and overall treatment costs. This trend has enhanced patient convenience and quality of life, eased capacity constraints in hospitals, and positioned outpatient CAR-T therapy as an increasingly viable treatment option for some patients.
Source data: Guidehouse Analysis of McKesson Compile Patient Ready Data (US)
European Commission Proposes Biotech Act to Strengthen EU Health Biotechnology
The European Commission published its proposal for a European Biotech Act in December 2025, as outlined in its 2024–2029 Political Guidelines. The Act seeks to create a supportive framework to accelerate the development of biotechnology products from research to market, while maintaining high safety and ethical standards. It represents a potential important milestone for ATMPs in Europe by seeking to better align regulation, investment, and patient access.
The Act combines regulatory improvements, targeted incentives, and funding measures to boost clinical trial activity, support ATMP centres of excellence, and enhance intellectual property protection, including a 12-month extension of supplementary protection certificates. It also aims to address Europe’s long-standing challenges in translating scientific excellence into commercial success, particularly in health biotechnology, where complex legislation and funding gaps had led to many developers focusing on other markets such as North America
Here are three main takeaways from the recently adopted Act:
- The Biotech Act seeks to address structural barriers limiting Europe’s ability to commercialise biotech innovation, including funding constraints and a declining share of global clinical trials.
- It introduces coordinated measures across seven core pillars, including strategic project designations, fast-track permitting, additional funding, and extended SPCs for qualifying biotech and ATMP products.
- The Act streamlines existing EU regulatory frameworks (including CTR, ATMP, and SoHO) through accelerated timelines, risk-based requirements, and regulatory sandboxes, aiming to reduce time to market while preserving high safety standards.
In parallel with the Biotech Act proposal, the Commission and the European Investment Bank Group announced BioTechEU, an initiative to mobilise €10 billion in public-private investment for the biotech and life sciences sectors in the next year.
ATMP Companies are Adapting Fast
To address the challenges within the ATMP sector, companies are increasingly required to adapt quickly and operate with greater agility in a rapidly evolving landscape. A growing number of developers are focusing on delivering best-in-class advanced therapies for indications with high unmet need and significant patient populations, including Parkinson’s disease, Multiple myeloma, Huntington’s disease, and others. This shift reflects both scientific progress and a broader commitment to addressing diseases where conventional treatment options remain limited.
In the early days of the ATMP sector, access to treatment was constrained by significant operational and infrastructure barriers, including a limited number of specialised treatment centres, Today, the sector is actively working to dismantle these barriers and expand patient access. For example, Carvykti, a personalised CAR-T cell therapy for adults with relapsed or refractory multiple myeloma, is now administered in an outpatient setting approximately 50% of the time. Developers are collaborating closely with regulatory agencies to enhance safety profiles and refine clinical trial designs in response to regulatory feedback, supporting more efficient and patient-centric development pathways. Heading into 2026, a significant number of regulatory decisions and submissions are planned for first half of the year and global clinical trials are gaining momentum.
HiTech Health is a leading European CDMO and service provider for ATMPs. We deliver end-to-end solutions from preclinical development through to GMP manufacturing and global patient supply. If you require support with the development and manufacturing of an ATMP, then contact us today.
Author: Paul Crozier – January 2026
References:
www.alliancerm.org/resources/?_resource_type=cell-gene-therapy-sector-data
https://alliancerm.org/wp-content/uploads/2026/01/SOTI-2026-Industry-Update.pdf
www.citeline.com/en/resources/q3-2025-gene-cell-and-rna-therapy-report
https://www.eesc.europa.eu/en/our-work/opinions-information-reports/opinions/biotech-act
https://www.businessnewsaustralia.com/articles/mesoblast-shares-hit-five-year-high-as-ryoncil-quarterly-sales-surge-60-per-cent-to-52m.html
https://ashpublications.org/blood/article-abstract/146/16/1897/546856/Remestemcel-L-rknd-Ryoncil-the-first-approved?redirectedFrom=fulltext