by Paul Crozier | Jan 22, | HiTech Health Latest News
By 2025, the global advanced therapy medicinal products (ATMP) sector, encompassing cell therapies, gene therapies, and genetically modified cell-based medicines, has progressed from early scientific success to clinical and commercial reality. Following a decade of rapid innovation, the ATMP sector is now characterised by a growing number of approved therapies, expanding therapeutic indications, and increasing global competition. The patient benefits of ATMPs are undeniable with a year-on-year growth in the number of patients who are benefitting from advanced therapies globally. Patients who previously had no treatment options are now being offered renewed hope through new innovative ATMPs. At the same time, the sector has faced commercial hurdles in 2025 including manufacturing and supply chain challenges, pricing and reimbursement scrutiny, and a renewed emphasis on demonstrating sustainable clinical and economic value at scale.
In 2025, 20 of the 30 largest biopharma companies by market cap are now investing in the development and commercialisation of ATMPs. Research conducted by Citeline reported ATMP sector funding of $11.1 Billion in 2025. The ATMP sector comprised approximately 18% of the value of all biotech venture financing in 2025, an increase in the reported 15% value report in 2024.
The Race to the Clinic: Asia Pacific Region Surpasses North America
In the past year, the Asia-Pacific region overtook North America for the first time in the number of ATMP clinical trials, signalling a significant shift in the global advanced therapy landscape. There were an estimated 916 clinical trials ongoing in North America in 2025, with 890 of these in the United States, while the Asia-Pacific region conducted approximately 990 trials, 716 of them in China. This growth in the Asia-Pacific region reflects the region’s growing focus on accelerating the translation of novel therapies from lab to clinic, supported by evolving government policies that are driving faster initiation of in-human studies. The trend highlights a broader intensifying competition to bring breakthrough ATMPs to patients worldwide.
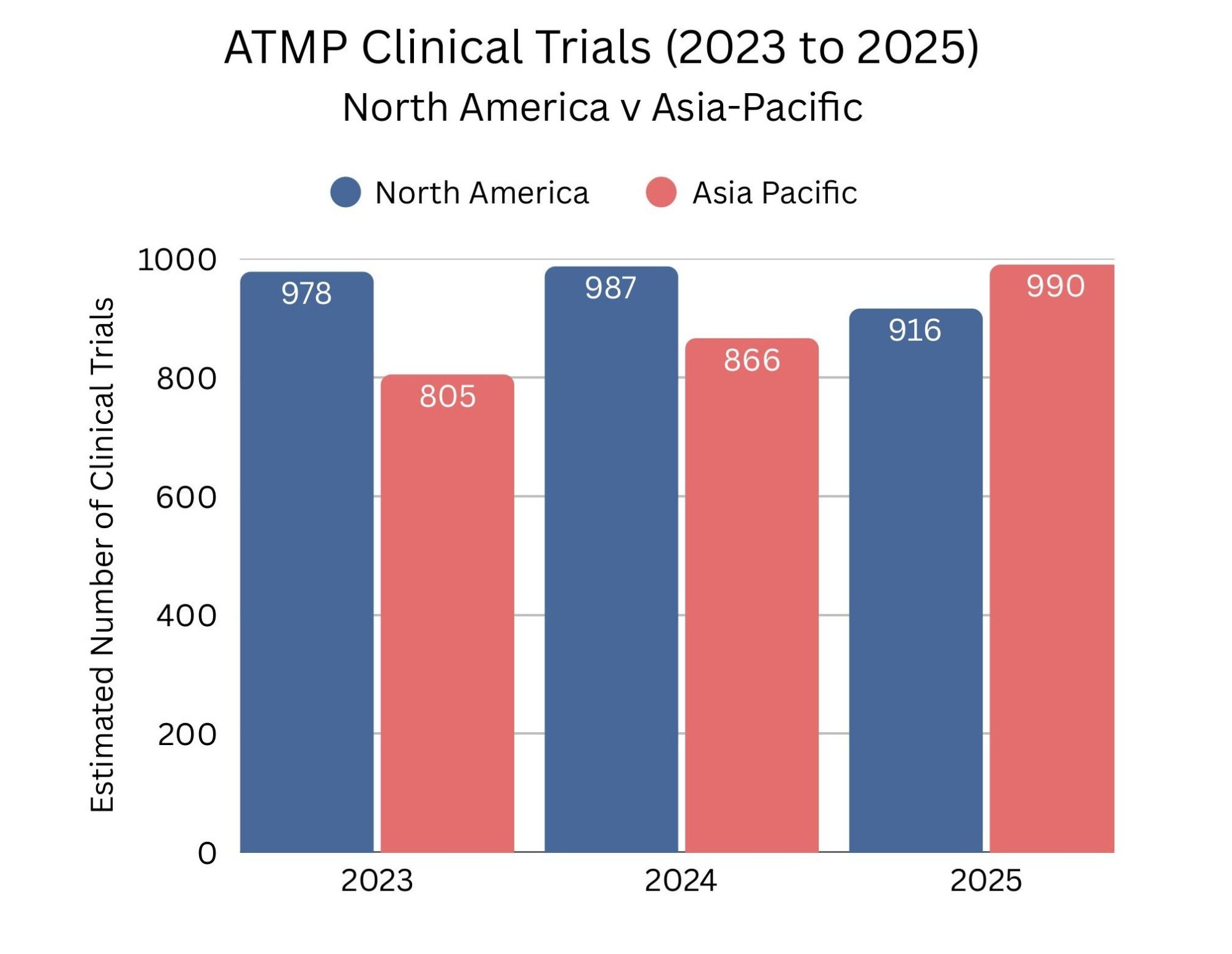
Source data: www.alliancerm.org
FDA and EMA Approvals in 2025
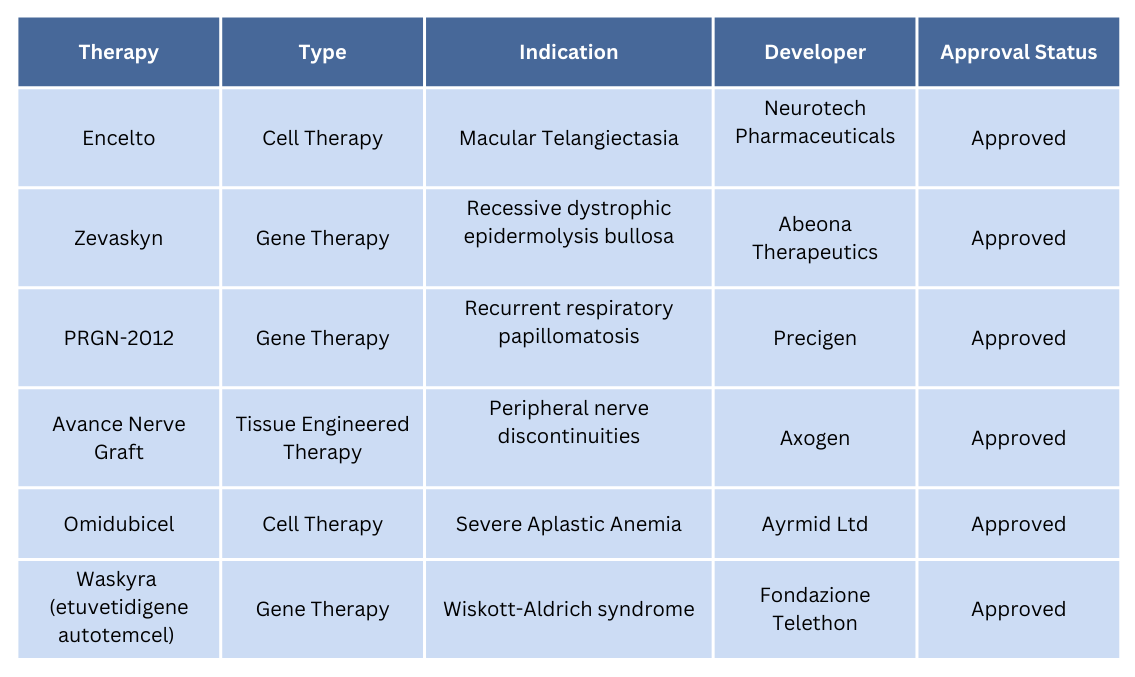
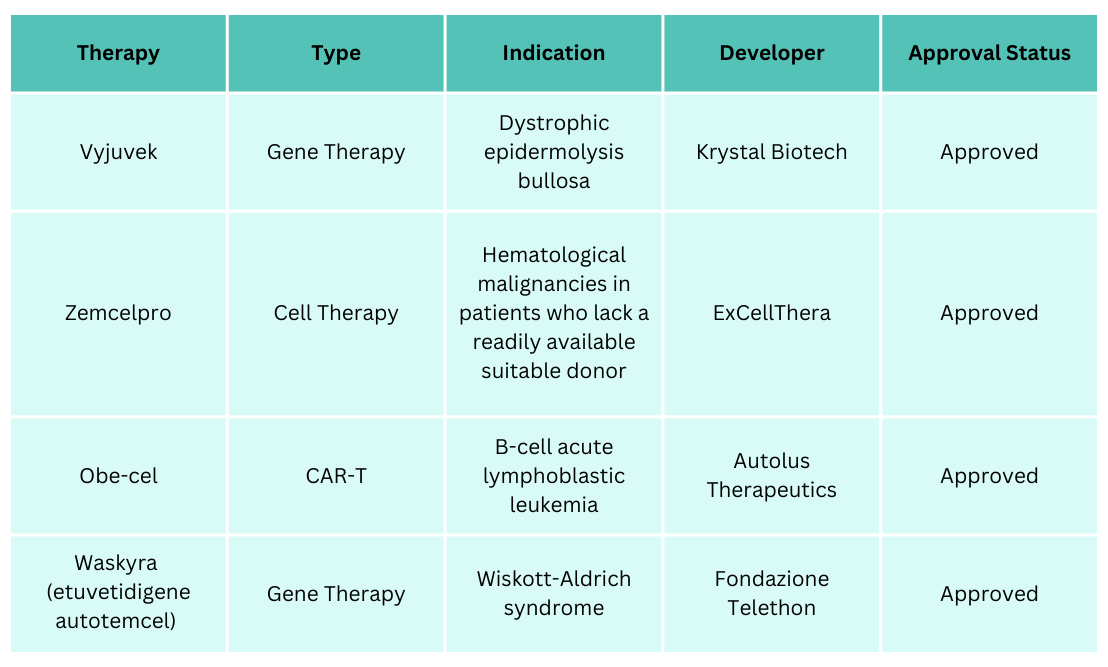
Over the past year, both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) granted several significant approvals for ATMPs. In total, the FDA approved six ATMPs, while the EMA approved four. A major milestone in 2025 was the approval of Fondazione Telethon’s etuvetidigene autotemcel (Waskyra) for Wiskott–Aldrich syndrome (WAS) by both the FDA and EMA. Waskyra is the first gene therapy approved for this rare disease, and Fondazione Telethon became the first non-profit organisation to achieve gene therapy approval. The tables below provide details on these newly authorised ATMPs.
ATMPs that were approved by the FDA in 2025
ATMPs that were approved by the EMA in 2025
Mesoblast’s Ryoncil became commercially available in the US in March 2025
At the end of 2024, Melbourne-based biotech Mesoblast secured FDA approval for Ryoncil, the first mesenchymal stromal cell (MSC) therapy to be approved by the FDA for any indication. Ryoncil is now an approved treatment for children aged two months and older, including adolescents, with steroid-refractory acute graft-versus-host disease (SRaGvHD), a life-threatening condition associated with high mortality. The therapy became commercially available in the US on 28th March 2025. Sales of Ryoncil reached US$13.2 million in the second quarter of 2025, increased to US$21.9 million in the third quarter (a 66% quarter-on-quarter increase), and rose further to US$35.1 million in the fourth quarter (approximately 60% growth over the previous quarter). This accelerating commercial performance reflects the growing number of patients benefiting from this MSC-based therapy.
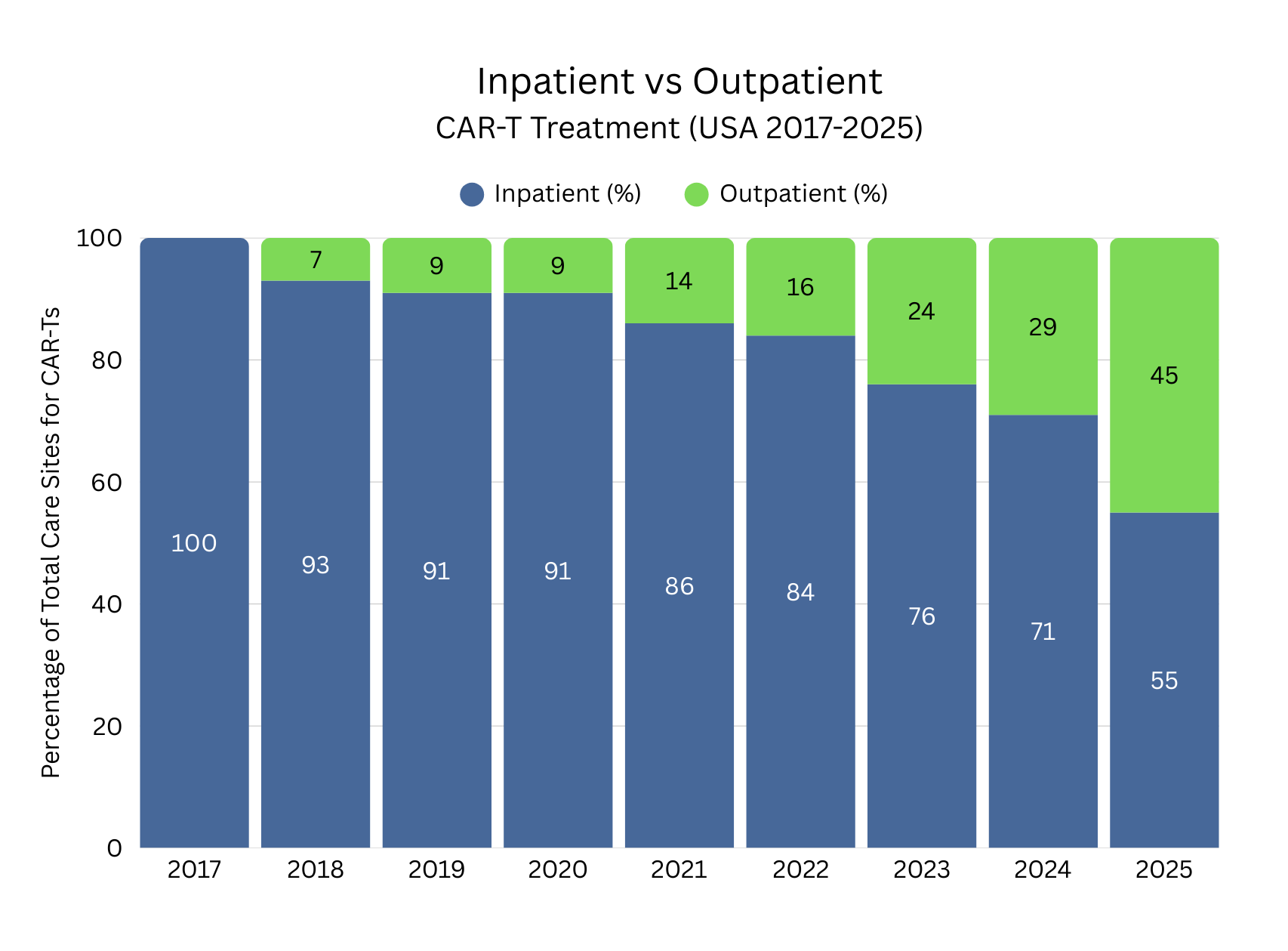
Significant Growth in Outpatient CAR-T Treatment
Among the positive developments has been the continued expansion of ATMP treatment access in outpatient settings. Outpatient CAR-T treatment has experienced significant growth in recent years, reflecting increasing understanding and clinical education on CAR-T administration, increased safety profiles, improved toxicity management and adapting healthcare infrastructure. Continuous improvements by developers and healthcare teams have made it possible to safely transition appropriate patients from inpatient to outpatient settings. Standardised monitoring protocols, remote symptom tracking, and rapid-response care pathways have increased clinician confidence in outpatient delivery while reducing hospital length of stay and overall treatment costs. This trend has enhanced patient convenience and quality of life, eased capacity constraints in hospitals, and positioned outpatient CAR-T therapy as an increasingly viable treatment option for some patients.
Source data: Guidehouse Analysis of McKesson Compile Patient Ready Data (US)
European Commission Proposes Biotech Act to Strengthen EU Health Biotechnology
The European Commission published its proposal for a European Biotech Act in December 2025, as outlined in its 2024–2029 Political Guidelines. The Act seeks to create a supportive framework to accelerate the development of biotechnology products from research to market, while maintaining high safety and ethical standards. It represents a potential important milestone for ATMPs in Europe by seeking to better align regulation, investment, and patient access.
The Act combines regulatory improvements, targeted incentives, and funding measures to boost clinical trial activity, support ATMP centres of excellence, and enhance intellectual property protection, including a 12-month extension of supplementary protection certificates. It also aims to address Europe’s long-standing challenges in translating scientific excellence into commercial success, particularly in health biotechnology, where complex legislation and funding gaps had led to many developers focusing on other markets such as North America
Here are three main takeaways from the recently adopted Act:
- The Biotech Act seeks to address structural barriers limiting Europe’s ability to commercialise biotech innovation, including funding constraints and a declining share of global clinical trials.
- It introduces coordinated measures across seven core pillars, including strategic project designations, fast-track permitting, additional funding, and extended SPCs for qualifying biotech and ATMP products.
- The Act streamlines existing EU regulatory frameworks (including CTR, ATMP, and SoHO) through accelerated timelines, risk-based requirements, and regulatory sandboxes, aiming to reduce time to market while preserving high safety standards.
In parallel with the Biotech Act proposal, the Commission and the European Investment Bank Group announced BioTechEU, an initiative to mobilise €10 billion in public-private investment for the biotech and life sciences sectors in the next year.
ATMP Companies are Adapting Fast
To address the challenges within the ATMP sector, companies are increasingly required to adapt quickly and operate with greater agility in a rapidly evolving landscape. A growing number of developers are focusing on delivering best-in-class advanced therapies for indications with high unmet need and significant patient populations, including Parkinson’s disease, Multiple myeloma, Huntington’s disease, and others. This shift reflects both scientific progress and a broader commitment to addressing diseases where conventional treatment options remain limited.
In the early days of the ATMP sector, access to treatment was constrained by significant operational and infrastructure barriers, including a limited number of specialised treatment centres, Today, the sector is actively working to dismantle these barriers and expand patient access. For example, Carvykti, a personalised CAR-T cell therapy for adults with relapsed or refractory multiple myeloma, is now administered in an outpatient setting approximately 50% of the time. Developers are collaborating closely with regulatory agencies to enhance safety profiles and refine clinical trial designs in response to regulatory feedback, supporting more efficient and patient-centric development pathways. Heading into 2026, a significant number of regulatory decisions and submissions are planned for first half of the year and global clinical trials are gaining momentum.
HiTech Health is a leading European CDMO and service provider for ATMPs. We deliver end-to-end solutions from preclinical development through to GMP manufacturing and global patient supply. If you require support with the development and manufacturing of an ATMP, then contact us today.
Author: Paul Crozier – January 2026
References:
www.alliancerm.org/resources/?_resource_type=cell-gene-therapy-sector-data
https://alliancerm.org/wp-content/uploads/2026/01/SOTI-2026-Industry-Update.pdf
www.citeline.com/en/resources/q3-2025-gene-cell-and-rna-therapy-report
https://www.eesc.europa.eu/en/our-work/opinions-information-reports/opinions/biotech-act
https://www.businessnewsaustralia.com/articles/mesoblast-shares-hit-five-year-high-as-ryoncil-quarterly-sales-surge-60-per-cent-to-52m.html
https://ashpublications.org/blood/article-abstract/146/16/1897/546856/Remestemcel-L-rknd-Ryoncil-the-first-approved?redirectedFrom=fulltext
by Paul Crozier | Jan 14, | HiTech Health Latest News
In the European Union (EU), the release of medicinal products for commercial supply is a highly regulated activity. Central to this release process is the Qualified Person (QP), a legally defined role under EU pharmaceutical legislation. The QP is responsible for ensuring that every batch supplied to the market meets the required standards of quality, safety, and compliance.
What Is a Qualified Person?
A Qualified Person is an individual named on a Manufacturer’s / Importer’s Authorisation (MIA) who meets specific education, training, and experience requirements to fulfil the role. The QP acts as the final accountable authority for batch certification prior to commercial distribution within the EU.
Key Responsibilities in Commercial Batch Release
Prior to certifying batches for market supply, the QP has to ensure:
- The medicinal product has been manufactured and tested in accordance with EU GMP and the approved Marketing Authorisation (MA).
- All manufacturing, testing, and packaging steps have been completed and documented in line with GMP requirements.
- Any deviations, changes, or out-of-specification results have been appropriately investigated and resolved.
- For imported products, manufacturing and quality controls conducted outside the EU are equivalent to EU GMP requirements.
Why the QP Role Matters?
The QP function is a critical component of the EU pharmaceutical regulatory system. It provides regulators and patients with assurance that medicinal products released to the market consistently meet EU standards, regardless of where they are manufactured. The role of the QP carries legal responsibility, with individual accountability to EU competent authorities for each certified batch. This responsibility underpins the assurance that product quality and patient safety consistently meet the highest regulatory standards.
For companies supplying medicinal products in the EU, access to an experienced QP team will help ensure timely and uninterrupted commercial supply. HiTech Health is a leading provider of QP services in the EU and also the UK. Our QP team have extensive knowledge of pharmaceuticals, biologics and Advanced Therapeutic Medicinal Products (ATMPs) including cell and gene therapies. We can help with all quality and compliance activities from your overall quality management system through quality audits and QMS remediation support.
Contact our team today and schedule a call to learn more about our QP services for the EU and UK markets.
by jgao | Sep 25, | HiTech Health Latest News
UK researchers said on Wednesday they have slowed the progression of the fatal neural condition Huntington’s disease for the first time with a groundbreaking new gene therapy.
Huntington’s disease causes nerve cells in the brain to decay over time. The disease affects a person’s movements, thinking ability and mental health. Huntington’s disease is rare and it’s often passed down through a changed gene from a parent. Medicines are presently available to help manage the symptoms of Huntington’s disease. However, these current medicines can’t prevent the physical, mental and behavioural decline caused by the disease.
The new study tested a new gene therapy, AMT-130, which is delivered through an injection directly into the brain. Researchers said that AMT-130 works by permanently introducing new functional DNA into a patient’s cells. Some of the patients who took part in early-stage clinical trials at University College London (UCL) saw the rate at which their condition developed reduced by 75 per cent after three years, according to uniQure, a gene therapy company based in the Netherlands and the United States. The new treatment is a type of gene therapy given during 12 to 18 hours of delicate brain surgery.
Read more here: https://www.bbc.com/news/articles/cevz13xkxpro
by jgao | Sep 4, | HiTech Health Latest News
Pharmaceutical and biotechnology companies are required to comply with strict regulations to ensure medicines are safe and effective. One key requirement for wholesaling activities is the Wholesale Distribution Authorisation (WDA). In the EU and UK, all wholesale distributors of medicinal products must possess an authorisation to engage in wholesaling activities related to medicinal products, safeguarding their quality, safety, and efficacy throughout the supply chain.
A WDA is required for any company that is involved in procuring, supplying, storing (storing medicinal products for over 72 hours), handling and distributing medicinal products within the EU or UK. This authorisation is a legal certification issued by national competent authorities to entities involved in the wholesale distribution of medicinal products. In the EU, the regulatory authority is the European Medicines Agency (EMA) and, in the UK, the regulatory authority is the Medicines and Healthcare products Regulatory Agency (MHRA).
This licence ensures that companies meet specific standards for procuring, supplying, storing, handling and exporting medicinal products to ensure quality and safety of the products is maintained throughout the supply chain. Companies without this licence are prohibited from legally distributing medicinal products to hospitals, pharmacies or other entities.
Furthermore, a WDA indicates that an organisation operates in compliance with Good Distribution Practice (GDP) guidelines. GDP assures that the medicinal products delivered to the end-user are safe, effective and high quality. By regulating this process, regulatory authorities aim to minimise risks related to product tampering, diversion, mishandling, contamination and the possible entry of falsified medicines into supply chains. Every WDA that is issued is legally required to have a named Responsible Person (RP) who is responsible for ensuring the conditions of the WDA are met.
Who is required to have a WDA?
Each medicinal product supply chain is unique and it is important to firstly map out the proposed business model to determine if a WDA is needed and what type of WDA is needed. Any entity who procures, holds, supplies or exports medicines to anyone other than the patient using the medicine needs a WDA:
- Procurement (purchase) of medicines for human use.
- Holding (storage) of medicinal products for human use.
- Supply (sale within the EEA) of medicinal products for human use, apart from to the public.
- Export (sale to outside the EEA) of medicinal products for human use, apart from to the public.
Without a WDA, these entities risk penalties, operational disruptions and reputational damage. Understanding whether your business operations fall under its scope is crucial to avoiding non-compliance.
Brokering of medicines is different to wholesaling, as it does not involve owning or physically handling medicines. It involves:
- Negotiation for procurement (purchase) or supply (sale) of medicines.
- Acting independently and on behalf of another legal or natural person.
Organisations that do not store medicines for more than 72 hours do not need a WDA. Furthermore, organisations that solely transport medicines do not need a WDA but are required to comply with GDP as it per pertains to transportation.
Require support with obtaining a WDA?
Navigating the requirements for obtaining a WDA can be difficult without the support of an experienced partner that is familiar with the regulatory requirements. HiTech Health has a team of Responsible Personnel (RPs) and Qualified Personnel (QPs) who are deeply experienced with working with a diverse range of life science companies to obtain a WDA. In addition to this, we provide contract RP services that allow our team to fulfil the role of RP on your behalf. This cost-effective solution includes end-to-end support from regulatory inspection through to oversight of wholesaling activities. Our team have led regulatory inspections on behalf of clients, delivering post-inspection support to address any inspection findings.
Contact our team today and schedule a call to find out how we can support your WDA application.
by jgao | Jul 23, | HiTech Health Latest News
The HiTech Health team has reviewed recent developments in the Advanced Therapeutic Medicinal Product (ATMP) sector during the first half of 2025. Below are some key updates from the industry so far:
Sector Summary:
- In Europe, the ATMP sector is on track to achieve 5-6 regulatory approvals in 2025, positioning the region to exceed the three approvals it saw in 2023 and 2024 combined.
- The US ATMP sector is presently expecting 5-6 approvals in 2025.
- There are at least nine ATMP developers aiming to submit regulatory applications for ATMPs in 2025. Further developers are targeting to file regulatory submissions in early 2026.
- The U.S could still potentially see four or more approvals for gene therapies to treat rare diseases in 2025, adding to the momentum of eight total approvals in 2023 and 2024.
- CAR-T cell therapies remained the most common technology used in the pipeline of genetically modified cell therapies (preclinical through to pre-registration).
- The first half of the year has seen significant clinical progress for therapies treating neurological disorders such as Parkinson’s Disease.
The global ATMP sector has seen significant progress in the first half of 2025, fuelled by breakthroughs in cell and gene research, encouraging clinical developments, and streamlined regulatory frameworks. The table below provides a snapshot of the estimated number of ongoing clinical trials, active ATMP developers, and investment activity within the sector.
| |
North America |
Europe |
Asia-Pacific |
H1 2025 Total |
| Number of Clinical Trials |
844 |
304 |
838 |
1,905 |
| Number of Developers |
770 |
453 |
750 |
2,070 |
| Investment |
$4.4B |
$0.8B |
$0.5B |
$5.0B |
Clinical Developments:
Below are some clinical developments across the ATMP sector:
- Epicrispr Biotechnologies announced in April that the FDA cleared its experimental epigenetic therapy for clinical trials in the United States, making it the first in-human trial to use epigenetic editing for this indication.
- Ensoma announced in May that its first-in-class in-vivo hematopoietic stem cell (HSC)-directed therapy to treat chronic granulomatous disease was approved for phase 1 clinical trials.
- Capsida Biotherapeutics announced in May that the FDA cleared its gene therapy for syntaxin-binding protein 1 developmental and epileptic encephalopathy, marking the first human clinical trial using an IV-administered blood-brain barrier crossing capsid.
- BrainStorm Cell Therapeutic’s Nurown received FDA clearance for phase 3b trials.
- XellSmart’s allogenic IPSC-derived cell therapy for Parkinson’s disease and ALS has been cleared for phase 1 clinical trials.
Regulatory and Policy Updates:
In June of this year, the FDA eliminated REMS requirements from approved CAR-T cell therapies and made other labelling updates, including reducing restricted driving time after the treatment from eight to two weeks and shortening the required stay near a healthcare facility post-treatment from four weeks to two. This decision was made in an effort to remove complex barriers affecting patient access.
European Commission Seeks to Drive ATMP Competitiveness
The European Commission announced in Q2 2025 the timely release of its European Life Sciences strategy, which aims to make Europe a global hub for the life sciences industry. The strategy outlines several positive steps to reduce barriers to the development of advanced therapies, such as European Centres of Excellence for ATMPs and funding for multi-country clinical trials.
Commercial Market Trends:
There are currently two cell and gene therapy blockbuster products ($1B+ in annual global sales). The table below gives an overview of the Q1 2025 revenue of the top grossing ATMPs:
| Therapy |
Q1 2025 Revenue |
Increase from Q1 2024 |
Blockbuster Status |
| Yescarta |
$383 million |
2% |
Achieved in 2022 |
| Elevidys |
$375 million |
179% |
Possible in 2025 |
| Carvykti |
$369 million |
135% |
On track for 2025 |
| Zolgensma |
$327 million |
10% |
Achieved in 2021 |
| Breyanzi |
$262 million |
148% |
On track for 2025 |
| Vyjuvek |
$88 million |
95% |
Possible by 2029 |
About HiTech Health:
We are a GMP-certified Contract Development and Manufacturing Organisation (CDMO) specialising in advanced therapies, including cell and gene therapies. Our team provide professional consulting services to support with product development, launch and supply across the advanced therapy, pharmaceutical, biotechnology, and medical device sectors.
Key Services or areas where HTH can help include:
Interested in Learning More?
Contact us today to arrange a call and discover how HiTech Health can support your organisation.
IE: +353-1-9631489 | UK: +44-20-30267419 | US: +1-857-3265835
Email: info@hitech-health.com
References
www.alliancerm.org
www.citeline.com
by jgao | Jul 1, | HiTech Health Latest News
The global clinical trials market is expected to nearly double to over £80 billion by 2032 and the United Kingdom remains one of the world’s leading destinations for conducting clinical research. Notably, the UK accounts for approximately 9.5% of all global cell and gene therapy clinical trials, highlighting its strength in the Advanced Therapy Medicinal Product (ATMP including cell and gene therapies) sector as well as broader pharmaceutical and biotechnology clinical research and development.
The consistent growth in the number of clinical trials conducted in the UK reflects its attractive research ecosystem, supported by strong infrastructure, a skilled workforce, and a progressive regulatory framework led by the Medicines and Healthcare products Regulatory Agency (MHRA). If you’re developing an Investigational Medicinal Product (IMP) and considering where to run your clinical trial, here are four reasons why you should consider the UK:
- Access to a Single, Publicly Funded Healthcare System – the NHS
The UK’s National Health Service (NHS) is the world’s largest publicly funded healthcare provider, delivering free care to approximately 69 million people and treating around 1.4 million patients every 24 hours. Furthermore, the NHS is deeply committed to engaging in clinical research and trials. The NHS Constitution pledges that all patients have the right to be informed about research studies they may be eligible for. In the past five years, 100% of NHS organisations have been research-active, and 75% have contributed to commercially sponsored clinical trials, making it a uniquely integrated and research-friendly healthcare system.
- Streamlined Regulatory Processes
The UK’s Combined Review allows for a single application to both the MHRA and Research Ethics Committee (REC), enabling faster approvals and reducing administrative burden. Recently introduced reforms to clinical trial regulation aim to make the process even more efficient and adaptable. In addition to this, the National Institute for Health and Care Research (NIHR) provides national infrastructure and support for high-quality clinical research, including feasibility assessments, site identification, and patient recruitment.
- Large and Diverse Population
With a population of approximately 69 million, the UK offers access to a large and ethnically diverse patient population, particularly in major urban centres like London. This diversity supports inclusive recruitment strategies and enables trial outcomes that are more generalisable, aligning with the increasing regulatory emphasis on population diversity from agencies including the FDA and EMA. HiTech Health have established relationships with clinical trial sites across the UK and our team have in-depth knowledge on what is required to initiate your clinical trial in the UK. The UK offers close collaboration between IMP developers, CDMOs, CROs, research institutes, and clinical trial sites, enabling efficient and coordinated study delivery.
- Access to a Skilled Workforce and Qualified Persons (QPs)
The UK is home to a highly skilled life sciences workforce, supported by world-class universities and a strong track record in innovative research. Clinical trial sponsors benefit from access to experienced scientists, clinical professionals, and Qualified Persons (QPs) who play a critical role in IMP batch certification and release.
While Brexit introduced separate regulatory pathways and licence requirements for the UK and EU, such as independent Manufacturer’s/Importer’s Authorisations (MIAs) and QP release, the industry has adapted well. At HiTech Health, we hold MIAs in both the UK and EU, and our QP teams operate across both regions, enabling seamless support for sponsors running trials in either or both jurisdictions. We are delighted to support a diverse portfolio of developers as they navigate the clinical trial landscape across Europe.
About HiTech Health (HTH)
We are a GMP-certified Contract Development and Manufacturing Organisation (CDMO) specialising in advanced therapies, including cell and gene therapies. Our team provide professional consulting services to support with product development, launch and supply across the advanced therapy, pharmaceutical, biotechnology, and medical device sectors.
Key Services or areas where HTH can help include:
Interested in Learning More?
Contact us today to arrange a call and discover how HiTech Health can support your clinical trial journey in the UK.
IE: +353-1-9631489 | UK: +44-20-30267419 | US: +1-857-3265835
Email: info@hitech-health.com
Author: Paul Crozier
Date: 1st July 2025