
by jgao | Jul 1, | HiTech Health Latest News
The global clinical trials market is expected to nearly double to over £80 billion by 2032 and the United Kingdom remains one of the world’s leading destinations for conducting clinical research. Notably, the UK accounts for approximately 9.5% of all global cell and gene therapy clinical trials, highlighting its strength in the Advanced Therapy Medicinal Product (ATMP including cell and gene therapies) sector as well as broader pharmaceutical and biotechnology clinical research and development.
The consistent growth in the number of clinical trials conducted in the UK reflects its attractive research ecosystem, supported by strong infrastructure, a skilled workforce, and a progressive regulatory framework led by the Medicines and Healthcare products Regulatory Agency (MHRA). If you’re developing an Investigational Medicinal Product (IMP) and considering where to run your clinical trial, here are four reasons why you should consider the UK:
- Access to a Single, Publicly Funded Healthcare System – the NHS
The UK’s National Health Service (NHS) is the world’s largest publicly funded healthcare provider, delivering free care to approximately 69 million people and treating around 1.4 million patients every 24 hours. Furthermore, the NHS is deeply committed to engaging in clinical research and trials. The NHS Constitution pledges that all patients have the right to be informed about research studies they may be eligible for. In the past five years, 100% of NHS organisations have been research-active, and 75% have contributed to commercially sponsored clinical trials, making it a uniquely integrated and research-friendly healthcare system.
- Streamlined Regulatory Processes
The UK’s Combined Review allows for a single application to both the MHRA and Research Ethics Committee (REC), enabling faster approvals and reducing administrative burden. Recently introduced reforms to clinical trial regulation aim to make the process even more efficient and adaptable. In addition to this, the National Institute for Health and Care Research (NIHR) provides national infrastructure and support for high-quality clinical research, including feasibility assessments, site identification, and patient recruitment.
- Large and Diverse Population
With a population of approximately 69 million, the UK offers access to a large and ethnically diverse patient population, particularly in major urban centres like London. This diversity supports inclusive recruitment strategies and enables trial outcomes that are more generalisable, aligning with the increasing regulatory emphasis on population diversity from agencies including the FDA and EMA. HiTech Health have established relationships with clinical trial sites across the UK and our team have in-depth knowledge on what is required to initiate your clinical trial in the UK. The UK offers close collaboration between IMP developers, CDMOs, CROs, research institutes, and clinical trial sites, enabling efficient and coordinated study delivery.
- Access to a Skilled Workforce and Qualified Persons (QPs)
The UK is home to a highly skilled life sciences workforce, supported by world-class universities and a strong track record in innovative research. Clinical trial sponsors benefit from access to experienced scientists, clinical professionals, and Qualified Persons (QPs) who play a critical role in IMP batch certification and release.
While Brexit introduced separate regulatory pathways and licence requirements for the UK and EU, such as independent Manufacturer’s/Importer’s Authorisations (MIAs) and QP release, the industry has adapted well. At HiTech Health, we hold MIAs in both the UK and EU, and our QP teams operate across both regions, enabling seamless support for sponsors running trials in either or both jurisdictions. We are delighted to support a diverse portfolio of developers as they navigate the clinical trial landscape across Europe.
About HiTech Health (HTH)
We are a GMP-certified Contract Development and Manufacturing Organisation (CDMO) specialising in advanced therapies, including cell and gene therapies. Our team provide professional consulting services to support with product development, launch and supply across the advanced therapy, pharmaceutical, biotechnology, and medical device sectors.
Key Services or areas where HTH can help include:
Interested in Learning More?
Contact us today to arrange a call and discover how HiTech Health can support your clinical trial journey in the UK.
IE: +353-1-9631489 | UK: +44-20-30267419 | US: +1-857-3265835
Email: info@hitech-health.com
Author: Paul Crozier
Date: 1st July 2025
by jgao | Apr 10, | HiTech Health Latest News
Dublin, Ireland, 10th April 2025 – HiTech Health, a Contract Development and Manufacturing Organisation (CDMO) specialising in advanced medicines, is delighted to communicate that the CanVas consortium led by HAON life sciences (a leading cell therapy biotech company) has received a €10.7 grant from the Disruptive Technology Innovation Fund (DTIF). The grant will help develop one of the first ever therapeutics for early brain injury by advancing Haon Life Science’s CanVas-001 therapy to the clinic.
CanVas-001 is an allogeneic investigational product containing two proprietary sourced cell types; ECFC (Endothelial Colony Forming Cells) and MSC (Mesenchymal Stromal Cells). CanVas-001 has demonstrated significant therapeutic benefit in large animal preclinical models of early brain injury. The CanVas platform is also developing drug candidates for adult neurodegenerative diseases.
The lead indication is Hypoxic Ischemic Encephalopathy (HIE), a devastating rare disease occurring when a baby’s brain does not receive enough oxygen or blood flow just before or shortly after birth. It is the principal cause of infant mortality and long-term neurologic disability, including accounting for a fifth of all cerebral palsy cases.
The CanVas consortium is a multidisciplinary team of experts, made up of the National Institute for Bioprocessing Research and Training (NIBRT) who will carry out analytical development, HiTech Health who will facilitate development and manufacture of CanVas-001 for clinic and University College Cork Infant Maternal and Child Health Research Centre, who will provide clinical development expertise in early brain injury and in particular, neonatal HIE.
About HiTech Health
HiTech Health (HTH) is a CDMO specialising in advanced therapies and small volume aseptic products. HiTech Health has process development laboratories and GMP manufacturing capabilities to support the development and manufacture of a diverse range of advanced medicines. HTH provides an end-to-end service including Qualified Person (QP), Responsible Person (RP), project and supply chain management services to ensure that products are manufactured, approved and distributed to clinical sites on time.
About DTIF
The Disruptive Technologies Innovation Fund (DTIF) is a €500 million challenge-based fund established under Project Ireland 2040. The objective of DTIF is invest in the development and deployment of disruptive technologies and applications on a commercial basis, driving collaboration between Ireland’s research base and industry to foster economic growth and innovation.
It is managed by the Department of Enterprise, Trade and Employment and administered by Enterprise Ireland.
About HIE
Hypoxic Ischemic Encephalopathy (HIE), a devastating rare disease occurring when a baby’s brain does not receive enough oxygen or blood flow just before or shortly after birth. It is a leading cause of infant mortality and long-term neurologic disability including accounting for a fifth of all cerebral palsy cases.
It is estimated one to three in every 1,000 births in Europe and the United States will be impacted by HIE resulting in 30,000 babies affected in these regions, each year. The incidence is far higher in low incomes countries with an estimate 1 million babies affected annually.
Contact us
by jgao | Apr 7, | HiTech Health Latest News
If you are planning on conducting clinical trials in the EU and are importing Investigational Medicinal Products (IMPs) then there are many factors to consider prior to clinical trial initiation. An IMP refers to a medicinal product that is being tested or used in a clinical trial to evaluate its efficacy, safety, or pharmacokinetics. The term is defined under the European Union’s Clinical Trials Regulation (EU) No. 536/2014 and applies to any medicine (including vaccines, biologics, cell or gene therapies) that is used in the context of clinical research. Commencing the journey to conduct clinical trials in the EU involves navigating both the specific country and general EU requirements, which is an intricate task.
Here are a few key considerations when importing IMPs into the EU:
1. Regulatory Authorisation
Prior to importing IMPs into the EU for use in clinical trials, you must submit a Clinical Trial Application (CTA) to the National Competent Authority (NCA) in the EU member state where the trial will take place. The CTA is a critical component of conducting clinical trials in the EU. If the proposed clinical trial involves multiple EU countries, the European Medicines Agency (EMA) and the Clinical Trials Regulation (CTR), which came into force in January 2022, provide a centralised process for multi-country trials. However, individual country regulations may still apply and it is recommended to discuss your clinical trial strategy with a regulatory expert.
2. Manufacturer’s /Importation Authorisation (MIA)
A Manufacturer’s/Importation Authorisation (MIA) is required if a company is involved in manufacturing and/or importation activities relating to human, veterinary or investigational medicinal product, within the EU as per GMP Annex 21. The term manufacturing and/or importation includes manufacturing, primary packaging, secondary packaging, physical importation of medicinal products or batch certification of medicinal products. To conduct clinical trials in the EU, it is not required that the Marketing Authorisation Holder (MAH)/Sponsor of the IMP hold their own MIA or hire an internal Qualified Person (QP) but they may partner with a company like HiTech Health who already hold MIAs in both the EU and UK and have a deeply experienced QP team in place. This can save you substantial time and money, thus enabling the MAH/Sponsor to commence clinical trials sooner.
3. Storage and Distribution Controls
Organisations are required to have appropriate controls in place for managing the storage and distribution of IMPs. Compliance with Good Distribution Practice (GDP) will help ensure safety, security and high product quality throughout the EU clinical supply chain. In some cases, IMPs need to be stored in a licenced depot or distribution centre in the EU, which must be approved for storing IMPs. Companies should consider how product temperature requirements will be maintained throughout the importation, storage and distribution activities.
HiTech Health holds both EU and UK MIAs as well as the ability to act as IOR for both the EU and the UK, allowing us to support global clients in overcoming complex challenges of importing IMPs and conducting clinical trials Europe as well as the UK. Our team have extensive hands-on experience with all aspects of importing IMPs into the EU/UK for clinical use including complex products such as cell or gene therapies, where direct to site importation is often required. By partnering with HiTech Health, our team guide you through the EU requirements and allow you to focus on other core business areas such as product development, clinical studies, and market access planning.
Schedule a call with our team today to learn more about our services and how we can support your organisation.
IE: +353-1-9631489 | UK: +44-20-30267419 | US: +1-857-3265835
Email: info@hitech-health.com
by jgao | Feb 5, | HiTech Health Latest News
Ireland has a strong track record of attracting major investment from the largest pharma companies in the world. The foundation of Ireland’s life sciences industry has been the small-molecule sector and has since developed expertise in biologics manufacturing. Ireland is home to nine of the world’s top 10 pharmaceutical companies and is the world’s third-largest pharmaceuticals exporter. The country has proven to be a successful gateway to the EU, UK, US, and other RoW markets, with companies availing of the operational, financial, and geographic benefits of setting up here or partnering with Irish-based companies. World-class regulation is an important element of that. Ireland has an exemplary compliance record with agencies like the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), both of which work closely with Ireland’s Health Products Regulatory Authority (HPRA).
The Growing Demand for Cell & Gene Therapies in Europe
There is rapidly growing diversity in therapeutic pipelines, including advancements in Cell & Gene Therapies Manufacturing. The global Advanced Therapeutic Medicinal Product (ATMP) market size was valued at USD 27.04 billion in 2022 and is expected to grow at a compound annual growth rate (CAGR) of 16.8% from 2023 to 2030. The emergence of cell and gene therapies has transformed the pharmaceutical industry and disease treatment landscape. It has opened new routes for the treatment of incurable diseases and several cancer types. The success of products such as Kymriah, Zolgensma, and others is the key factor driving the market.
Why Ireland for ATMP and Cell and Gene Therapy Manufacturing?
Ireland is actively supporting the growing field of cell and gene therapy, and companies are capitalising on the list of benefits that Ireland affords them. The Irish government is committed to making the country a global leader for the development, manufacture, supply, and adoption of advanced therapies, including cell and gene therapies and novel vaccines, through strong investment in world-class training and academic development, an innovative research base, and excellent clinical trials infrastructure. This synergy between industry and academia, bolstered by strong government support, has contributed to Ireland’s competitive edge in attracting and retaining talent in advanced therapies. For developers of cell and gene therapies, Ireland offers research and development incentives, access to an extensive network of clinical trial sites, and the availability of providers who have exemplary track records with regulatory authorities.
Key benefits of establishing ATMP manufacturing in Ireland include:
- World-class regulatory environment ensuring compliance with EMA, FDA, and HPRA standards.
- Strategic location offering seamless access to the EU, UK, and global markets.
- Tax incentives and R&D grants for biopharmaceutical companies.
- Highly skilled workforce with specialised expertise in advanced therapy manufacturing.
- Strong collaboration between industry and academia, fostering innovation in advanced therapies.
Comprehensive ATMP Manufacturer and Cell & Gene Therapies CDMO in Europe
HiTech Health is an ATMP Manufacturer in Europe, positioned at the forefront of Ireland’s Cell & Gene Therapies CDMO sector. Our EU GMP-certified manufacturing facility supports the development and manufacturing of advanced therapies from a strategic European base. HiTech Health was the first Irish company to secure a licence to contract manufacture cell and gene therapies on behalf of international clients. As a leading ATMP Manufacturer in Europe, our team of experts facilitates the transformation of a promising new therapy idea from initial development to the GMP manufacture of a drug to treat patients in a clinical environment, helping to accelerate the journey of advanced therapies towards commercialisation. Our GMP facility is equipped with cutting-edge technology designed to advance the development, production, and testing of sterile formulations.
HiTech Health also provides consulting services to biopharma companies to develop, launch, and support products. We hold Manufacturer’s / Importer’s Authorisations (MIAs) in both the EU and UK to seamlessly support companies with batch importation and release for clinical and commercial use. HiTech Health is working with early-stage developers to provide end-to-end cell & gene therapy CDMO services as well as batch approval and supply to clinical sites. For companies located outside Europe, we can support navigating the landscape for advanced medicines across all EU countries. Unlike the traditional pharmaceutical CDMO model, HiTech Health also has a logistics team to support the shipping and storage of time and temperature-sensitive therapies. Our expertise as a Cell & Gene Therapies CDMO allows us to provide comprehensive solutions tailored to the needs of biotech innovators and pharmaceutical companies.
Our full range of services to accelerate cell and gene therapy commercialisation, covering but not limited to:
- GMP manufacturing of ATMPs for clinical and commercial use.
- Regulatory consulting to support EMA, FDA, and HPRA approvals.
- Batch importation and release for the EU and UK markets under our Manufacturer’s / Importer’s Authorisations (MIAs).
- End-to-end CDMO services for early-stage developers.
- Logistics solutions for time and temperature-sensitive therapies.
- Clinical trial support, ensuring seamless supply chain management.
If you’d like to connect with a member of our team to learn more about our cell and gene therapy manufacturing services, please email info@hitech-health.com.
References
Enterprise Ireland – Pharma
IDA Ireland – Biopharma
Grand View Research – ATMP Market
HiTech Health – Cell and Gene Therapy Facility Launch
HiTech Health – Key Considerations for Selecting a Cell and Gene Therapy CDMO
by jgao | Jan 21, | HiTech Health Latest News
HiTech Health are delighted to have an article entitled ‘Cell and Gene Therapy Manufacturing Considerations for Early Stage Companies’ published in distinguished publications, cellandgene.com and outsourcedpharma.com. The article should be of benefit to companies who are looking for a partner to develop and manufacture their clinical products.
Read the full article by clicking here: https://www.cellandgene.com/doc/cell-gene-therapy-manufacturing-considerations-for-early-stage-companies-0001
HiTech Health is a leading European CDMO and service provider for ATMPs. Learn more about our cell and gene therapy services by clicking here
Schedule a call with our team today to learn more about our services and how we can support your organisation.
IE: +353-1-9631489 | UK: +44-20-30267419 | US: +1-857-3265835
Email: info@hitech-health.com
by Paul Crozier | Dec 11, | HiTech Health Latest News
Over the past decade, the regenerative medicine landscape has undergone an extraordinary evolution with the continuous growth of Advanced Therapeutic Medicinal Products (ATMPs). ATMPs are medicines for human use that are based on genes, tissues or cells. They treat the root cause of diseases and disorders by altering, augmenting, repairing, replacing, or regenerating organs, tissues, cells, genes and metabolic processes in the body. This means that they can offer potentially groundbreaking new opportunities to address unmet medical needs.
10 years ago, in 2014, the ATMP sector was starting to grow but several hurdles including development, manufacturing and regulatory challenges and uncertainties needed to be addressed. Back then the field was still in its infancy, with only a few approved therapies and a rapidly growing number of early stage development programmes. The FDA had approved a small number of cell therapies, including several cord blood-based products like Hemacord and Allocord. However, there were no FDA-approved gene therapies in 2014.1
So much has changed in a positive way for patients. In 2024, the ATMP sector has expanded significantly, with therapies now available for a diverse range of medical conditions and they have become more accessible. There have been major improvements in the development, manufacturing and supply chains of ATMPs that have helped more patients receive these lifesaving and life changing medicines. There are now multiple gene therapies on the market for rare genetic disorders, such as Spinal Muscular Atrophy and Duchenne Muscular Dystrophy.
The sector has made significant advancements in 2024, driven by positive regulatory approvals, breakthroughs in scientific research, manufacturing optimisations and progress in market access and pricing. ATMPs have already saved and improved many lives, for example, a treatment for spinal muscular atrophy, Zolgensma, has been delivered to over 3,700 children globally. The sector has shown robustness and promising foundations for continued sector growth in 2025 and beyond. Developers are not only focused on proving that therapies can work but, are placing increased emphasis on refining their effectiveness, reducing side effects, expanding their indications and addressing market access and cost challenges.2
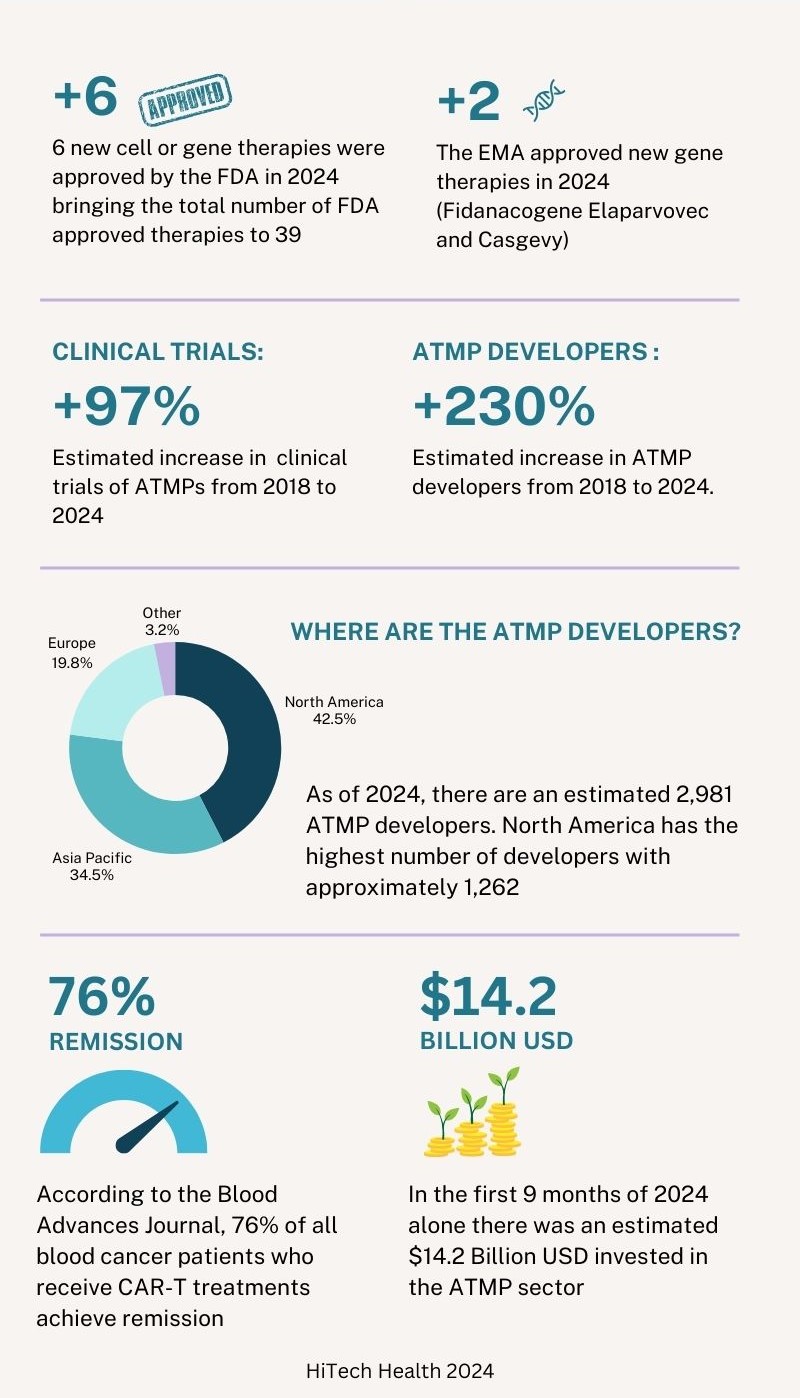
At the end of 2024, there were 39 cell and gene therapies approved in the United States by the FDA. Of the 6 new therapies approved by the FDA in 2024, 3 were gene therapies and 3 were cell therapies. In 2024 there have also been 2 new gene therapies approved by the European Medicines Agency (EMA). In addition to this, the FDA, EMA and other regulatory agencies are actively reviewing submissions made in 2024 and it is expected that a number of approval decisions will be made in the first quarter of 2025. The table below shows the therapies that were approved by the FDA and EMA in 2024.3
| Therapy |
Type |
Indication |
Developer |
Approval Status |
| Lifileucel |
Cell Therapy |
Metastatic Melanoma |
Iovance Biotherapeutics |
FDA approved 16 February 2024 |
| Lenmeldy |
Gene Therapy |
Metachromatic Leukodystrophy |
Orchard Therapeutics |
FDA approved 18 March 2024 |
| Fidanacogene Elaparvovec |
Gene Therapy |
Hemophilia B |
Pfizer |
FDA approved 27 April 2024
EMA approved 25 July 2024 |
| Tecelra |
Cell Therapy |
Advanced Synovial Sarcoma |
Adaptimmune Therapeutics |
FDA approved 1 August 2024 |
| Obecabtagene Autoleucel |
Cell Therapy |
B-Cell Acute Lymphoblastic Leukaemia |
Autolus Therapeutics |
FDA approved 8 November 2024 |
| Kebilidi |
Gene Therapy |
Aromatic L-Amino Acid Decarboxylase Deficiency |
PTC Therapeutics |
FDA approved 14 November 2024 |
| Casgevy |
Gene Therapy |
Sickle cell Disease and Beta-Thalassemia |
Vertex Pharmaceuticals and Crispr Therapeutics |
EMA approved 13 February 2024
(Previously approved by the FDA in late 2023 with an expanded label approval on 16 January 2024) |
Six years ago, in 2018, there were approximately 1000 clinical trials and 900 developers across the ATMP sector. By the end of the third quarter of 2024, the number of clinical trials has increased by approximately 97% in 6 years with an estimated 1,968 clinical trials. The number of developers has increased by approximately 230% during the same period to 2,981 developers. Of this estimated number of developers, 1,262 are based in North America, 1,036 are based in Asia Pacific, 587 are based in Europe and 96 are in other regions. Compared to the data in 2023, ‘other regions’ outside North America, Asia Pacific and Europe have seen the largest growth with an estimated yearly increase of 12.9% followed by Asia Pacific (12%), North America (6.6%) and Europe (3.3%).3
Similar to 2023, the majority of the clinical trials still involve cell-based therapies, accounting for approximately 70% of all trials. Oncology remains to be the most prevalent indication for ATMPs. Cancer patients, especially those who relapse, often must endure a series of arduous treatments using current standard of care chemotherapy and radiotherapy. CAR-T therapies have shown a reduction in collateral damage to healthy cells and can minimise side effects compared to traditional treatments. According to the Blood Advances Journal, 76% of all blood cancer patients who receive CAR-T treatments achieve remission. For B cell Lymphoma, overall survival is almost 9 times higher than the standard of care. Patients who receive Yescarta, a therapy used to treat B cell lymphoma, are 21% less likely to require subsequent treatment (insert reference: Vanderbilt University).3 4
Looking into the investments made in the ATMP sector, there was an estimated $13.3 billion USD invested 6 years ago in 2018 (excluding mergers and acquisitions (M&A) and grants). In the first 9 months of 2024, there has been a total estimated investment of $14.2 billion. At the time of writing, the data for the final 3 months of 2024 is not yet available but it is projected that the total annual investment will surpass $17 billion USD. There have been fluctuations in total investment over the last several years but the sector has shown robustness and positive investor sentiment. Across all regions, the largest type of investment is in the form of venture financing. North America has consistently seen the highest levels of investment since the industry was first developed.3
In Europe, in January 2022, Regulation (EU) 2021/2282 on health technology assessment (HTA) came into effect. The regulation aims to be applied in EU Member States by next month on the 12th January 2025. The HTA introduces an evidence‑based process that will allow the competent EU and national authorities to determine the effectiveness of new or existing health technologies, including cell and gene therapies. From 2025, all cell and gene therapies will undergo a single EU assessment of the value they add to patients and healthcare systems, aiming to end the need for 27 individual reviews. Companies will also meet jointly with the European Medicines Agency and Europe’s HTA coordinating group to discuss and align on the optimal clinical trial designs that deliver data, not only on safety and efficacy but also on added value to patients and healthcare systems.5
The regulation of ATMPs has progressed significantly in both the US, Europe and Asia Pacific regions over the last several years. In the US, the FDA has defined more transparent pathways for ATMPs, supported by the Regenerative Medicine Advanced Therapy designation, which facilitates expedited review and development support. The EMA has refined its guidelines for ATMPs, publishing a joint action plan with the European Commission to offer more streamlined processes for developers. There are opportunities for improvements in the regulatory pathways and we expect that the coming years will yield further changes. Developers are seeking more harmonised international regulations to help them navigate approvals across multiple regions more effectively. Notably, the regulatory agencies have also responded to the growing need for post-market surveillance, recognising the long-term nature of many ATMPs and the requirement for ongoing monitoring of their safety and efficacy.
2024 continues the trend of significant advances in the development and approval of new ATMPs which, ultimately, is great news for patients. The number of therapies receiving regulatory approval remains high with several applications still under review by the agencies. While there are challenges for ATMPs in manufacturing, pricing, and safety, the significant increase in the number of clinical trials demonstrates the potential of these medicines to enhance and transform patients’ lives. The outlook for the sector remains strong, with continued investment, technological innovation, and regulatory support propelling the industry forward in 2025 and beyond.
Hitech Health is a leading European CDMO and service provider for ATMPs. If you require support with the development and manufacturing of an ATMP, schedule a meeting with our team by emailing info@hitech-health.com.
References:
1 – https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products
2 – https://www.novartis.com/news/media-releases/novartis-presents-new-data-safety-and-efficacy-zolgensma-including-maintained-and-improved-motor-milestones-older-and-heavier-children-sma
3 – https://alliancerm.org/data/
4 – https://www.hematology.org/newsroom/press-releases/2023/a-promising-outlook-car-t-cells-improve-patient-quality-of-life
5 – https://health.ec.europa.eu/health-technology-assessment/implementation-regulation-health-technology-assessment/joint-clinical-assessments_en





